iPS Cell Advanced Characterization and Development Team, BioResource Research Center, RIKEN, 3-1-1 Koyadai, Tsukuba, Ibaraki, 305-0074, Japan.
School of Integrative and Global Majors, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki, 305-8577, Japan.
Hum Cell. 2024 Nov 13;38(1):18. doi: 10.1007/s13577-024-01147-x.
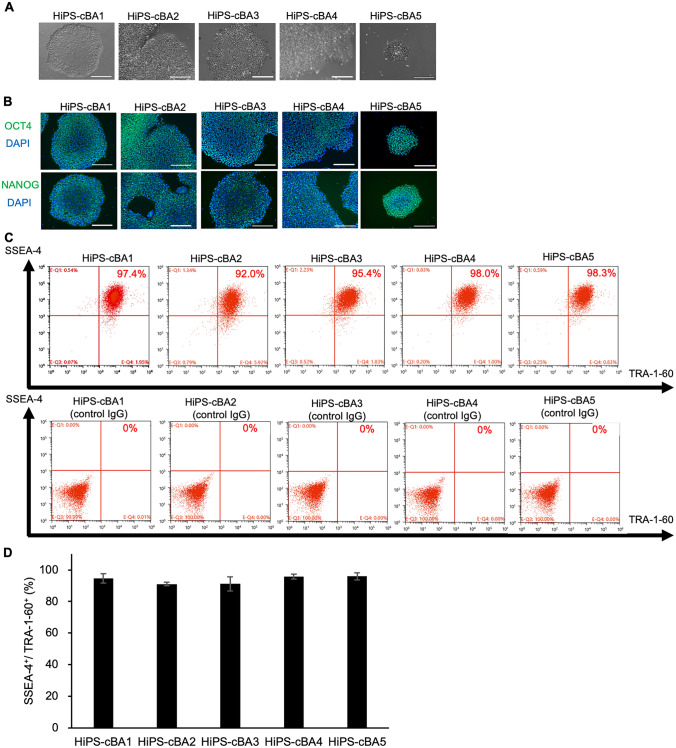
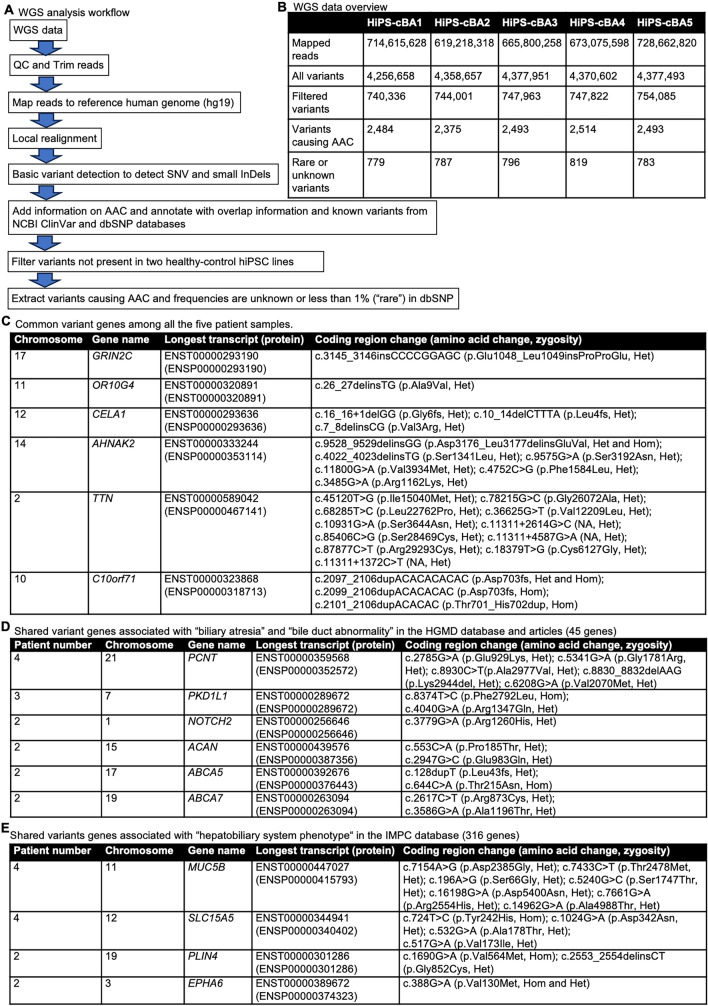
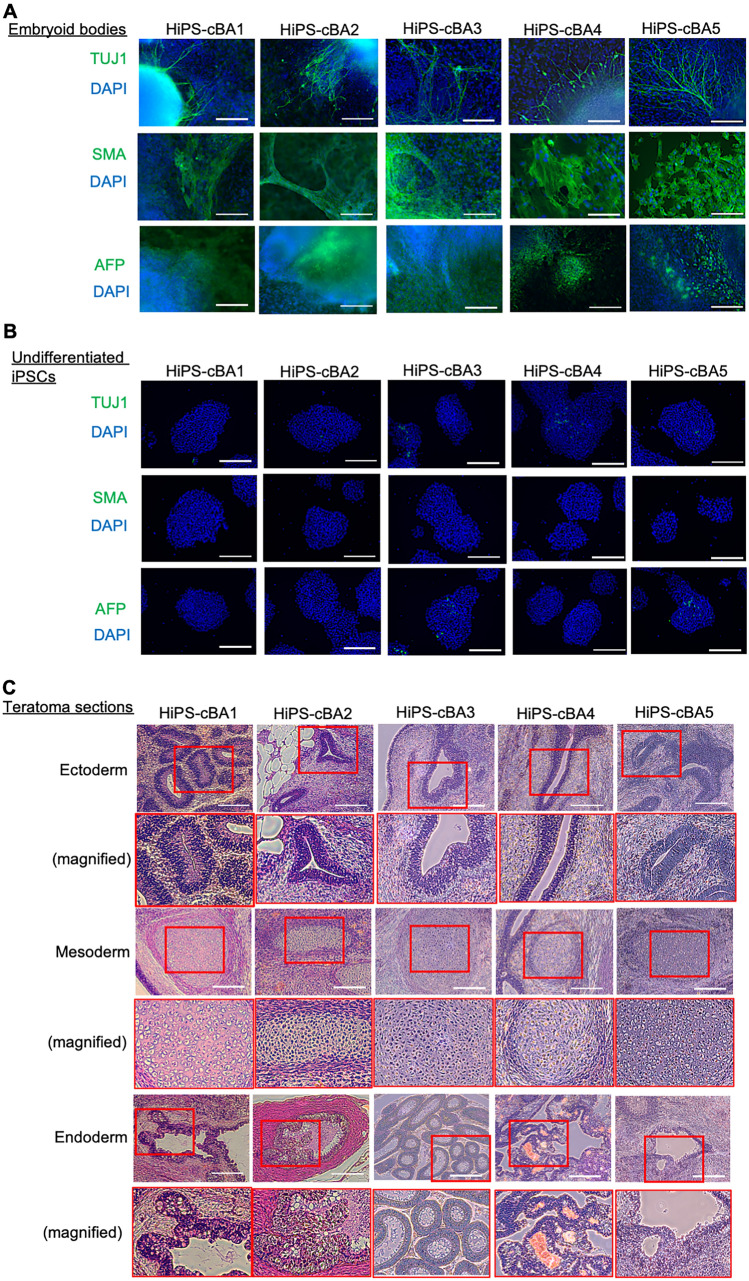
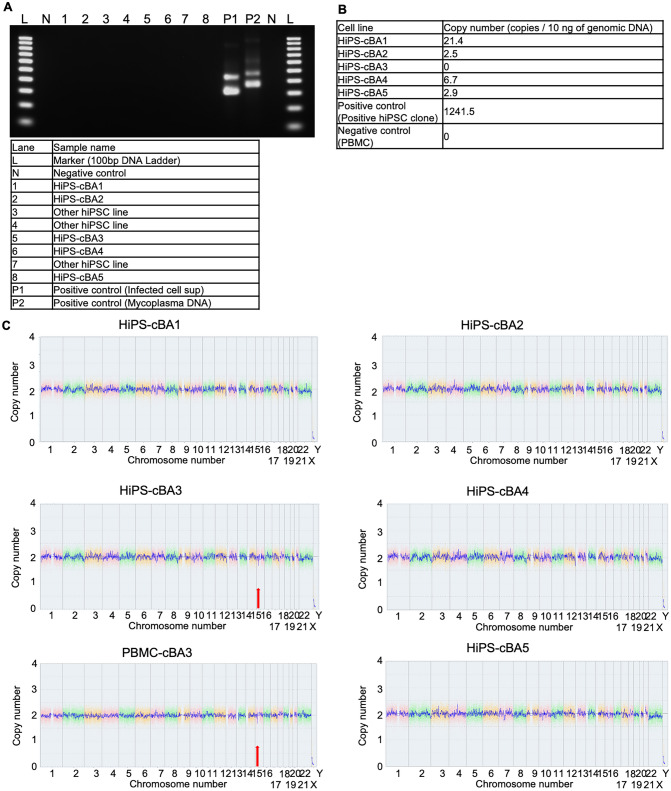
Biliary atresia (BA), resulting from abnormal development of the liver's internal or external bile ducts, can lead to liver damage and potentially fatal cirrhosis. Type I cystic biliary atresia is a relatively uncommon, but clinically significant variant of BA. It is critical to develop experimental models of BA to examine the etiology and pathogenesis, which remain elusive, and to develop future therapeutics. Here, we have successfully generated a panel of human induced pluripotent stem cells (hiPSCs) from five Japanese patients carrying type I cystic BA. These hiPSC lines exhibited characteristics of self-renewal and pluripotency. These cells held normal karyotypes mostly, but one of them carried hemizygous deletions, the clinical significance of which is unknown yet. Whole genome sequence analysis indicated that some of the mutations or single nucleotide polymorphisms (SNPs) commonly found in these patients are related to hepatobiliary abnormality. Given the limited understanding of the molecular pathogenesis of cystic BA, attributed to unknown factors of genetic and environmental causes, these cellular resources will be instrumental in replicating disease phenotypes and in advancing novel therapies for this disease.
先天性胆道闭锁(BA)是由于肝脏内部或外部胆管的异常发育引起的,可导致肝脏损伤,并可能导致致命的肝硬化。Ⅰ型囊性胆道闭锁是一种相对罕见但具有临床意义的 BA 变体。开发 BA 的实验模型对于研究其病因和发病机制至关重要,这些机制仍不清楚,同时也有助于开发未来的治疗方法。在这里,我们已经成功地从五名患有Ⅰ型囊性 BA 的日本患者中生成了一组人诱导多能干细胞(hiPSC)。这些 hiPSC 系表现出自我更新和多能性的特征。这些细胞大多具有正常的核型,但其中一个细胞携带半合子缺失,其临床意义尚不清楚。全基因组序列分析表明,这些患者中常见的一些突变或单核苷酸多态性(SNP)与肝胆异常有关。鉴于对囊性 BA 的分子发病机制的了解有限,这归因于遗传和环境因素的未知因素,这些细胞资源将有助于复制疾病表型,并为这种疾病推进新的治疗方法。