Department of Oncology, The Sidney Kimmel Comprehensive Cancer Center.
Department of Medicine.
J Pediatr Gastroenterol Nutr. 2019 Jan;68(1):56-63. doi: 10.1097/MPG.0000000000002187.
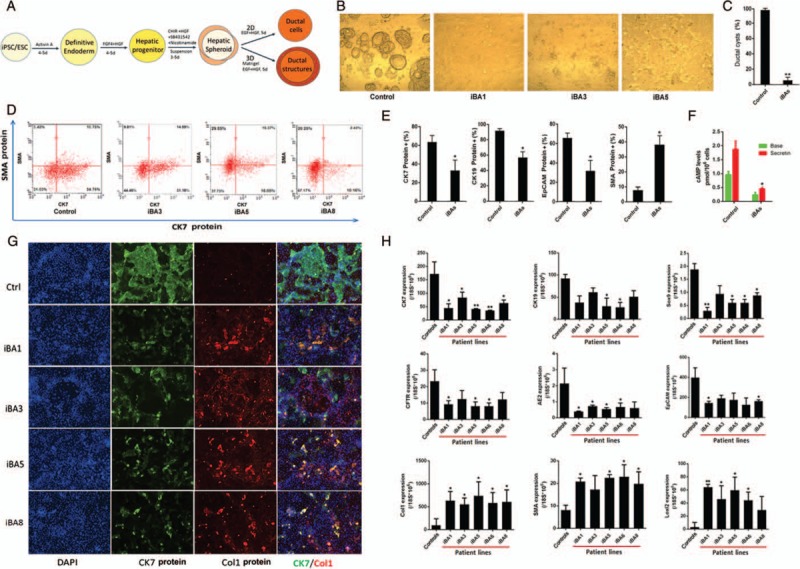
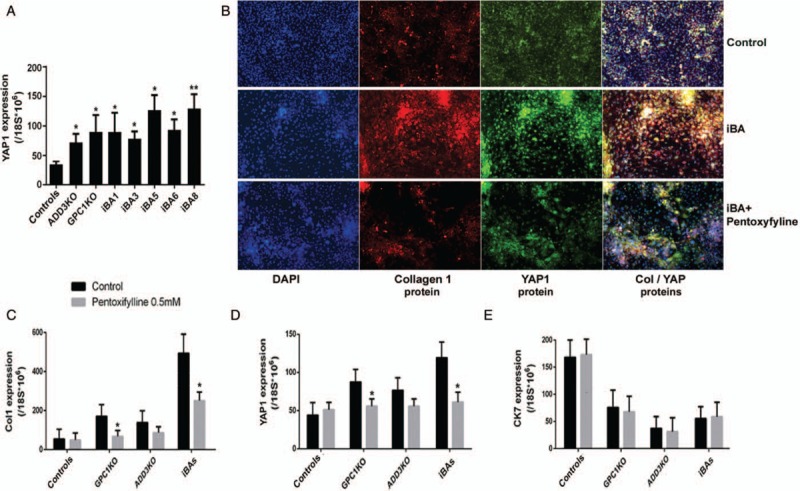
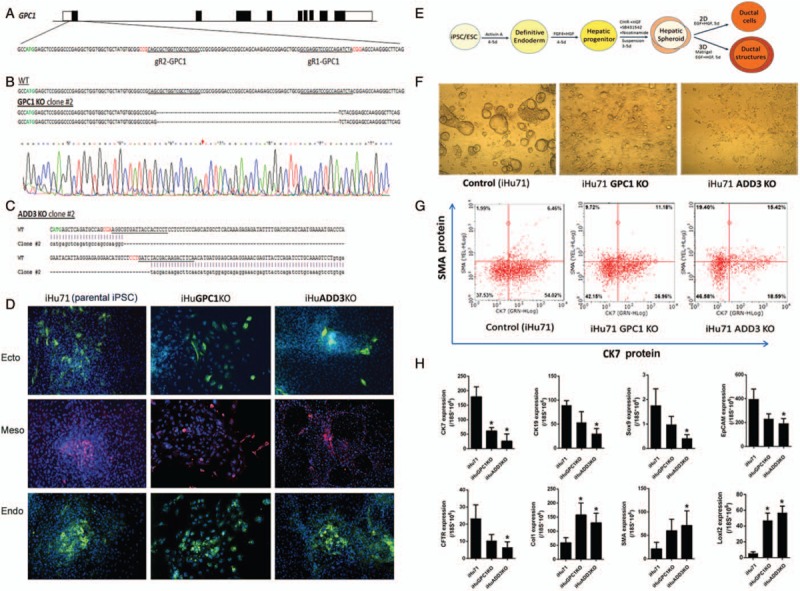
Biliary atresia (BA) is the most common cause of pediatric end-stage liver disease and the etiology is poorly understood. There is no effective therapy for BA partly due to lack of human BA models. Towards developing in vitro human models of BA, disease-specific induced pluripotent stem cells (iPSCs) from 6 BA patients were generated using non-integrating episomal plasmids. In addition, to determine the functional significance of BA-susceptibility genes identified by genome-wide association studies (GWAS) in biliary development, a genome-editing approach was used to create iPSCs with defined mutations in these GWAS BA loci. Using the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system, isogenic iPSCs deficient in BA-associated genes (GPC1 and ADD3) were created from healthy iPSCs. Both the BA patient-iPSCs and the knock out (KO) iPSCs were studied for their in vitro biliary differentiation potential. These BA-specific iPSCs demonstrated significantly decreased formation of ductal structures, decreased expression of biliary markers including CK7, EpCAM, SOX9, CK19, AE2, and CFTR and increased fibrosis markers such as alpha smooth muscle actin, Loxl2, and Collagen1 compared to controls. Both the patient- and the KO-iPSCs also showed increased yes-associated protein (YAP, a marker of bile duct proliferation/fibrosis). Collagen and YAP were reduced by treatment with the anti-fibrogenic drug pentoxifylline. In summary, these BA-specific human iPSCs showed deficiency in biliary differentiation along with increased fibrosis, the 2 key disease features of BA. These iPSCs can provide new human BA models for understanding the molecular basis of abnormal biliary development and opportunities to identify drugs that have therapeutic effects on BA.
先天性胆道闭锁(BA)是小儿终末期肝病的最常见原因,其病因尚未完全阐明。由于缺乏人类 BA 模型,部分原因是缺乏有效的治疗方法。为了开发 BA 的体外人类模型,使用非整合的附加体质粒从 6 名 BA 患者中生成了疾病特异性诱导多能干细胞(iPSC)。此外,为了确定全基因组关联研究(GWAS)中确定的 BA 易感性基因在胆管发育中的功能意义,使用基因组编辑方法在这些 GWAS BA 基因座中创建了具有定义突变的 iPSC。使用成簇规律间隔短回文重复序列(CRISPR)/Cas9 系统,从健康的 iPSC 中创建了与 BA 相关基因(GPC1 和 ADD3)缺陷的同基因 iPSC。对 BA 患者-iPSC 和敲除(KO)iPSC 进行了体外胆管分化潜能研究。与对照相比,这些 BA 特异性 iPSC 显示出明显减少的导管结构形成,减少了胆管标记物的表达,包括 CK7、EpCAM、SOX9、CK19、AE2 和 CFTR,以及增加了纤维化标记物,如α平滑肌肌动蛋白、Loxl2 和 Collagen1。与对照相比,患者和 KO-iPSC 还显示出增加的 yes 相关蛋白(YAP,胆管增殖/纤维化的标志物)。用抗纤维化药物己酮可可碱治疗可减少胶原蛋白和 YAP。总之,这些 BA 特异性人 iPSC 显示出胆管分化不足,同时伴有纤维化增加,这是 BA 的 2 个关键疾病特征。这些 iPSC 可为了解异常胆管发育的分子基础以及为 BA 提供具有治疗效果的药物提供新的人类 BA 模型。