Subbanna Mythili S, Winters Matthew J, Örd Mihkel, Davey Norman E, Pryciak Peter M
Department of Biochemistry and Molecular Biotechnology, University of Massachusetts Chan Medical School, Worcester, MA 01605, USA.
University of Cambridge, Cancer Research UK Cambridge Institute, Robinson Way, Cambridge CB2 0RE, UK.
bioRxiv. 2024 Nov 1:2024.10.30.621084. doi: 10.1101/2024.10.30.621084.
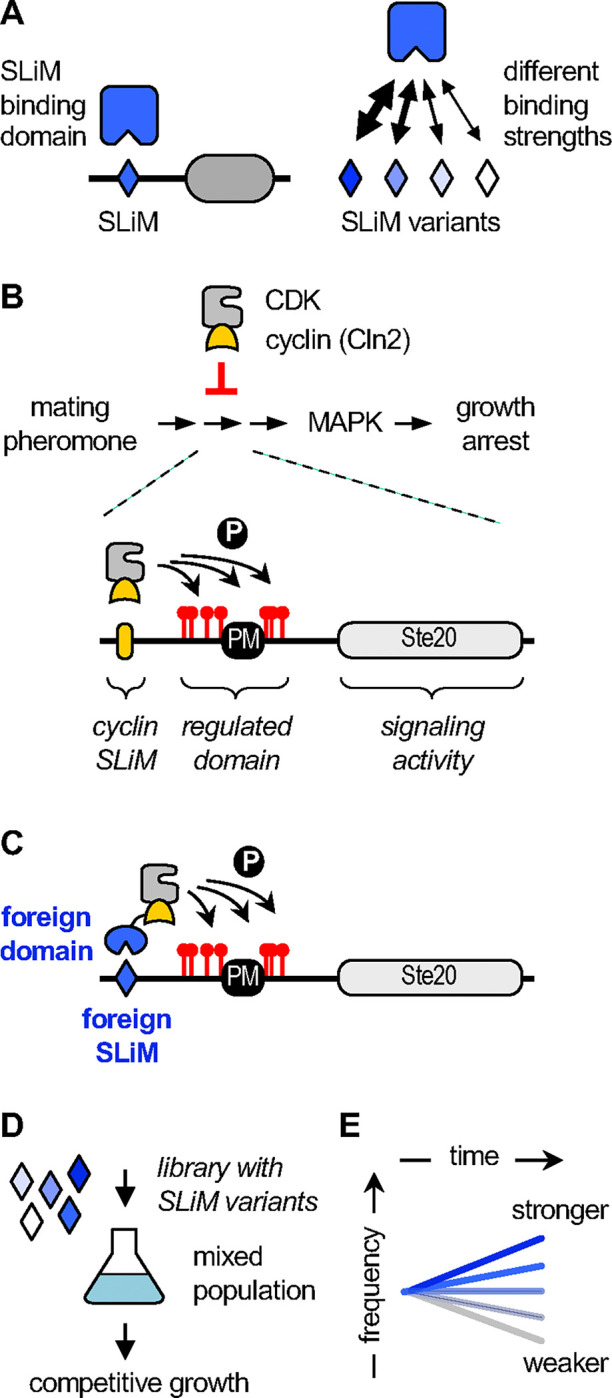
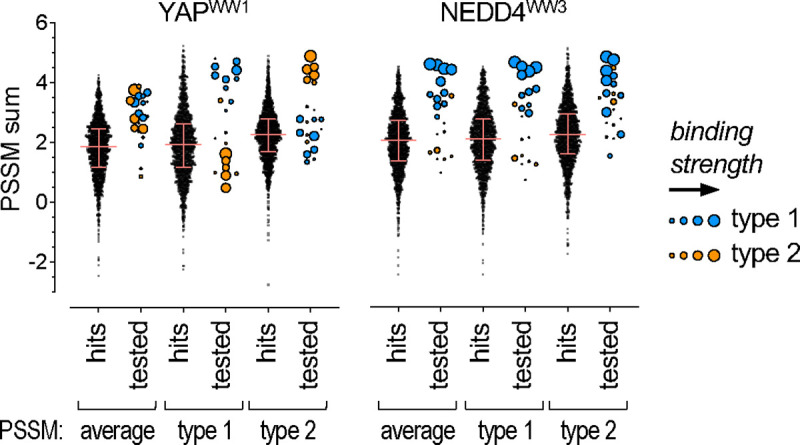
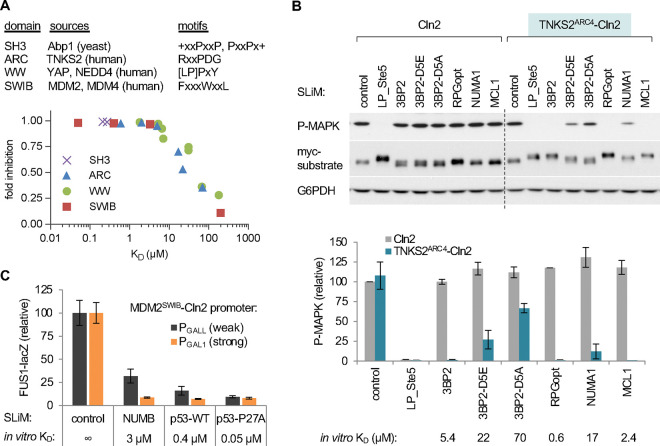
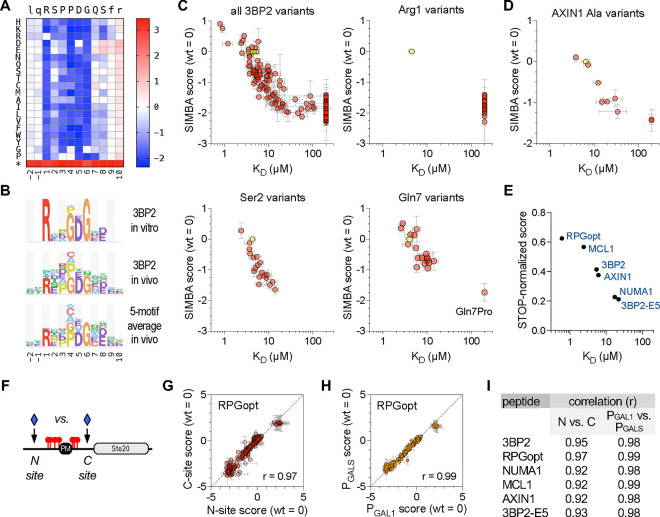
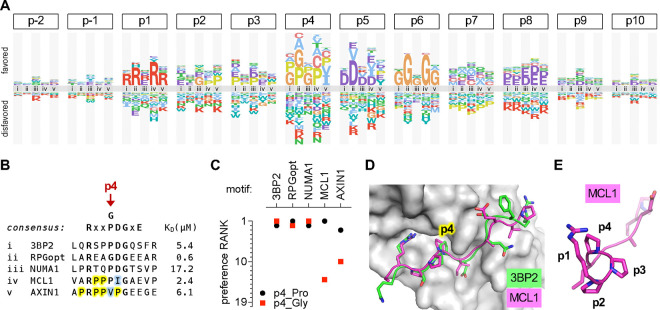
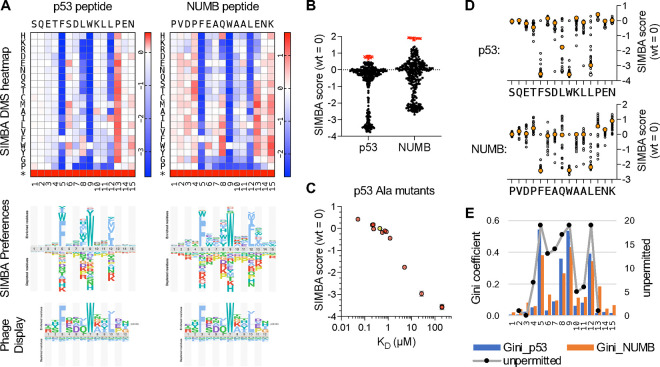
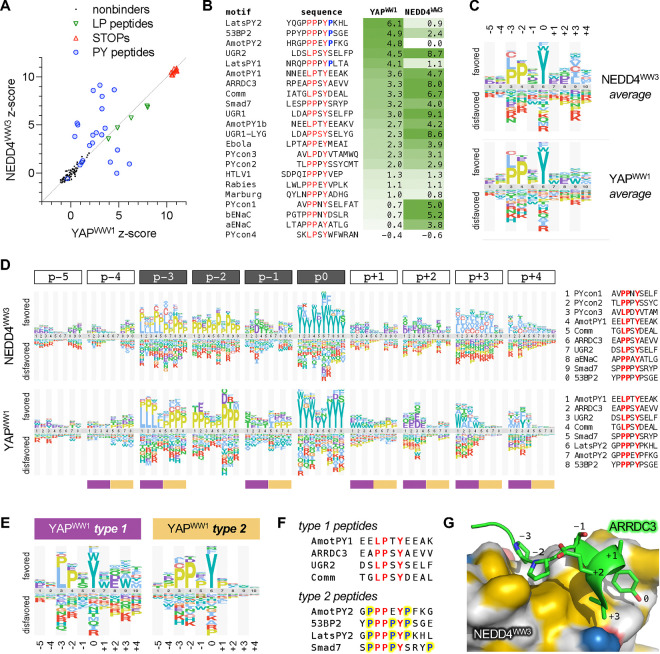
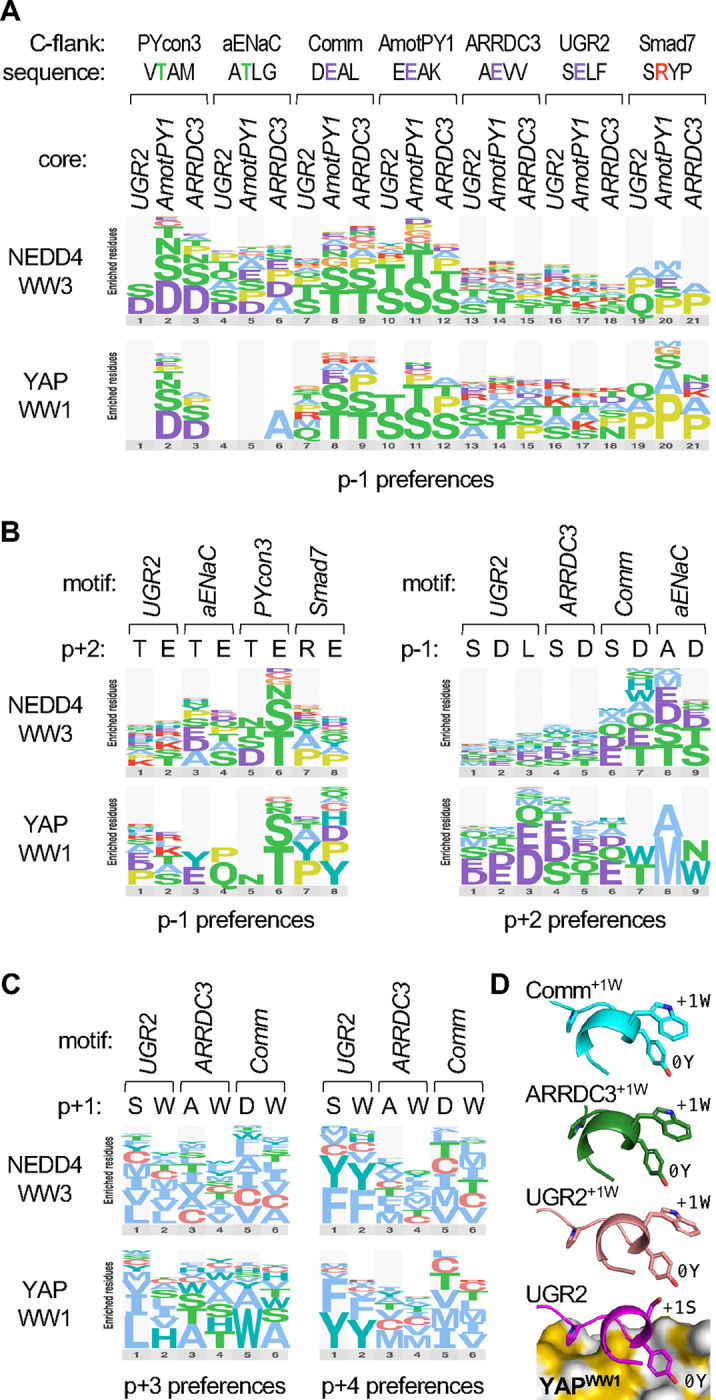
Transient protein-protein interactions play key roles in controlling dynamic cellular responses. Many examples involve globular protein domains that bind to peptide sequences known as Short Linear Motifs (SLiMs), which are enriched in intrinsically disordered regions of proteins. Here we describe a novel functional assay for measuring SLiM binding, called Systematic Intracellular Motif Binding Analysis (SIMBA). In this method, binding of a foreign globular domain to its cognate SLiM peptide allows yeast cells to proliferate by blocking a growth arrest signal. A high-throughput application of the SIMBA method involving competitive growth and deep sequencing provides rapid quantification of the relative binding strength for thousands of SLiM sequence variants, and a comprehensive interrogation of SLiM sequence features that control their recognition and potency. We show that multiple distinct classes of SLiM-binding domains can be analyzed by this method, and that the relative binding strength of peptides in vivo correlates with their biochemical affinities measured in vitro. Deep mutational scanning provides high-resolution definitions of motif recognition determinants and reveals how sequence variations at non-core positions can modulate binding strength. Furthermore, mutational scanning of multiple parent peptides that bind human tankyrase ARC or YAP WW domains identifies distinct binding modes and uncovers context effects in which the preferred residues at one position depend on residues elsewhere. The findings establish SIMBA as a fast and incisive approach for interrogating SLiM recognition via massively parallel quantification of protein-peptide binding strength in vivo.
瞬时蛋白质-蛋白质相互作用在控制细胞动态反应中起着关键作用。许多例子涉及球状蛋白质结构域与被称为短线性基序(SLiMs)的肽序列结合,这些基序在蛋白质的内在无序区域中富集。在这里,我们描述了一种用于测量SLiM结合的新型功能测定法,称为系统细胞内基序结合分析(SIMBA)。在这种方法中,外源球状结构域与其同源SLiM肽的结合通过阻断生长停滞信号使酵母细胞增殖。SIMBA方法的高通量应用涉及竞争性生长和深度测序,可快速定量数千种SLiM序列变体的相对结合强度,并全面探究控制其识别和效力的SLiM序列特征。我们表明,多种不同类别的SLiM结合结构域都可以通过这种方法进行分析,并且体内肽的相对结合强度与其体外测量的生化亲和力相关。深度突变扫描提供了基序识别决定因素的高分辨率定义,并揭示了非核心位置的序列变异如何调节结合强度。此外,对结合人端锚聚合酶ARC或YAP WW结构域的多个亲本肽进行突变扫描,确定了不同的结合模式,并揭示了上下文效应,即一个位置上的优选残基取决于其他位置的残基。这些发现确立了SIMBA作为一种通过体内蛋白质-肽结合强度的大规模平行定量来探究SLiM识别的快速而精确的方法。