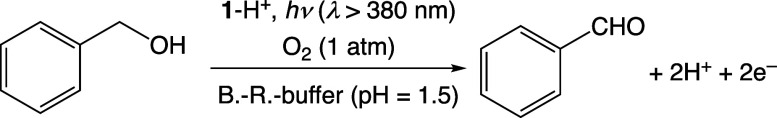Ishizuka Tomoya, Nishi Taichiro, Namura Nanase, Kotani Hiroaki, Osakada Yasuko, Fujitsuka Mamoru, Shiota Yoshihito, Yoshizawa Kazunari, Kojima Takahiko
Department of Chemistry, Faculty of Pure and Applied Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8571, Japan.
SANKEN (The Institute of Scientific and Industrial Research), Osaka University, Ibaraki, Osaka 567-0047, Japan.
J Am Chem Soc. 2024 Dec 4;146(48):33022-33034. doi: 10.1021/jacs.4c09962. Epub 2024 Nov 19.
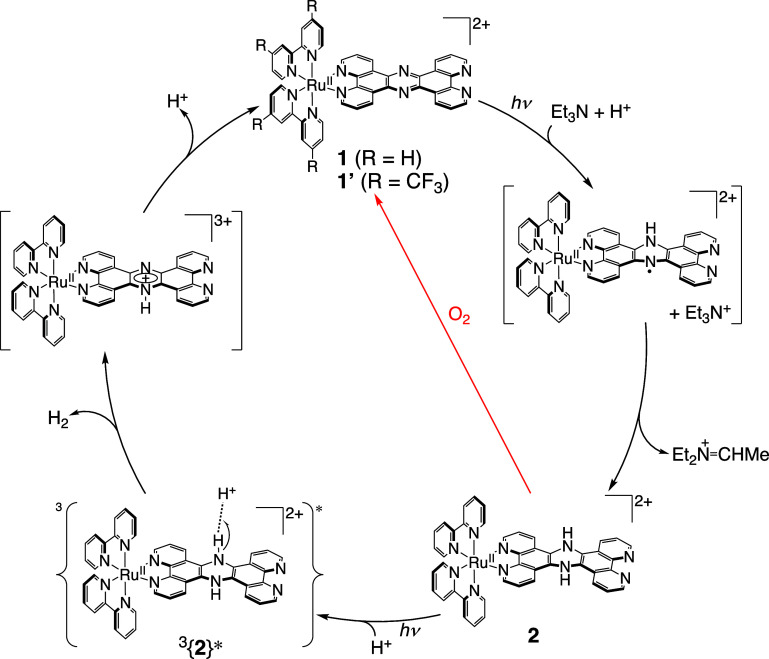
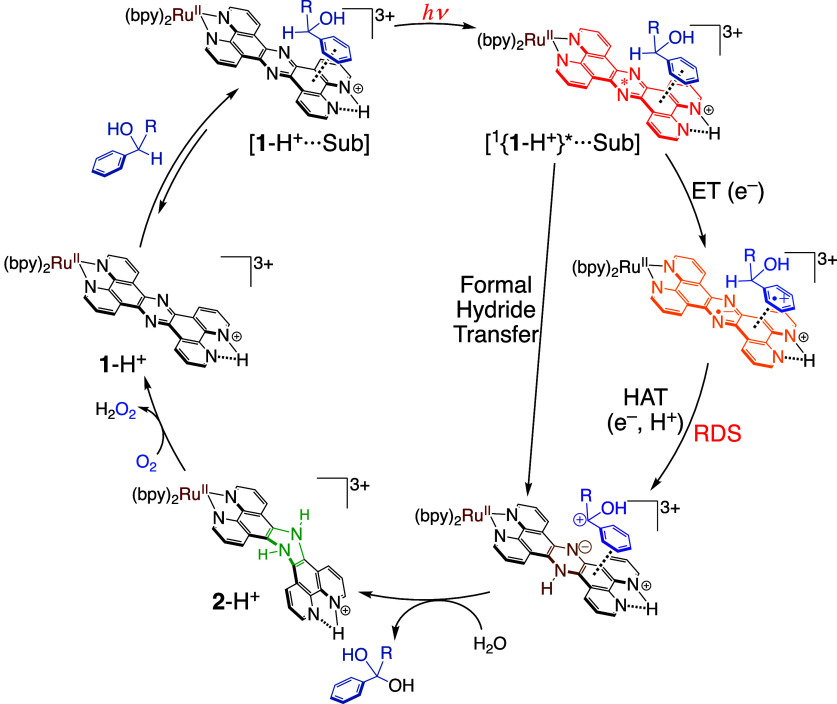
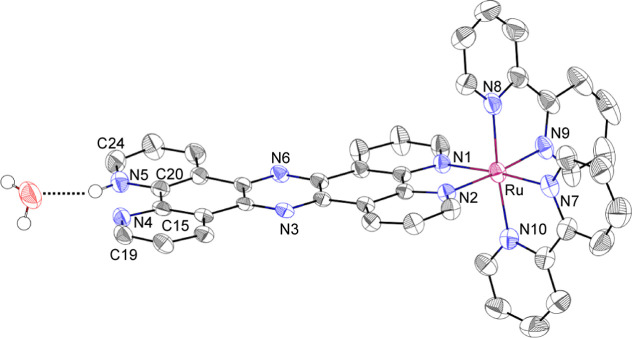
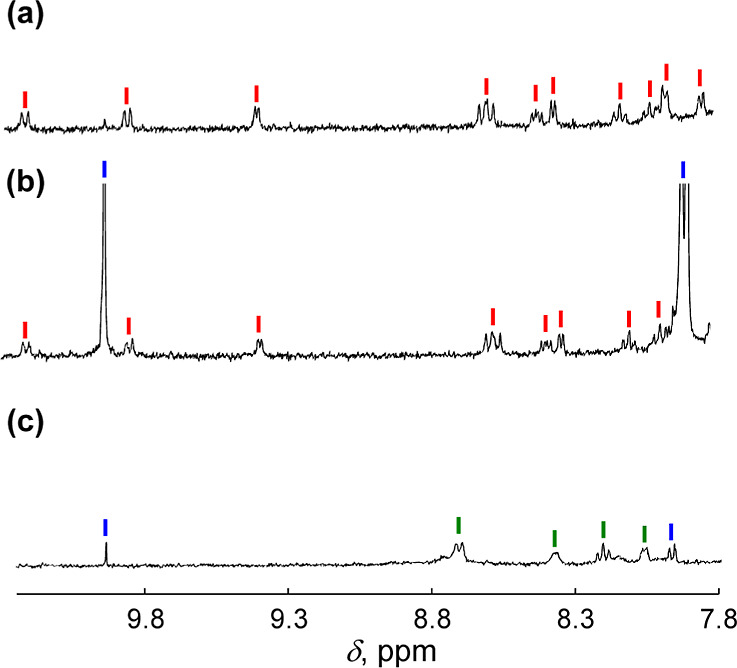
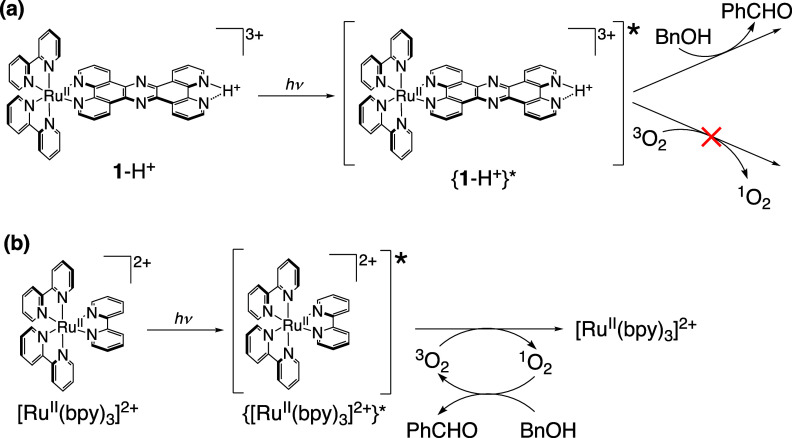
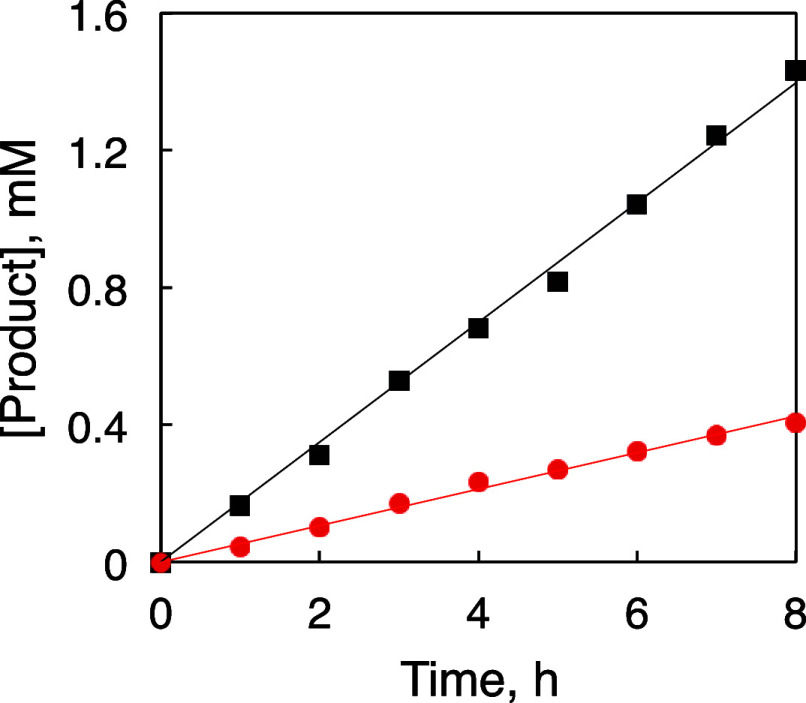
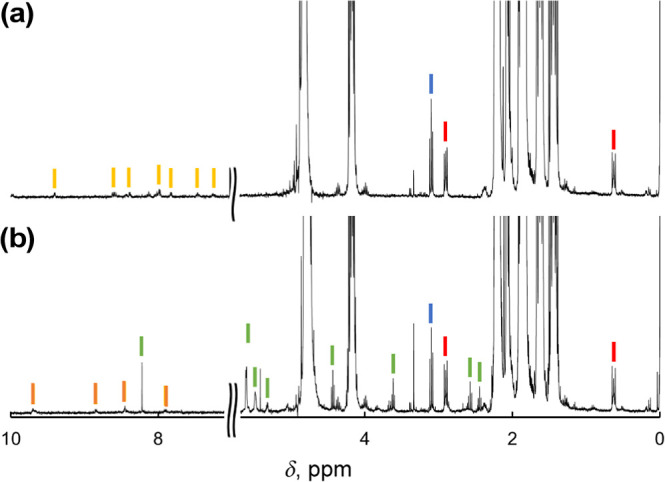
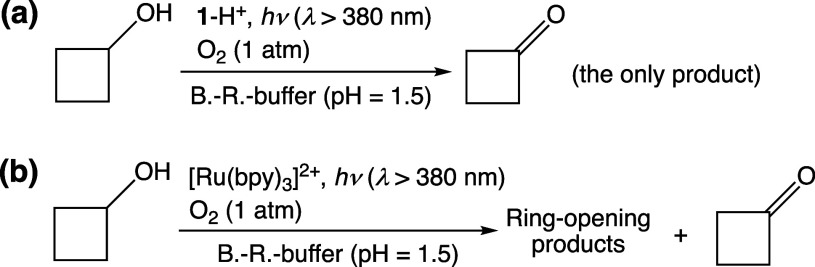
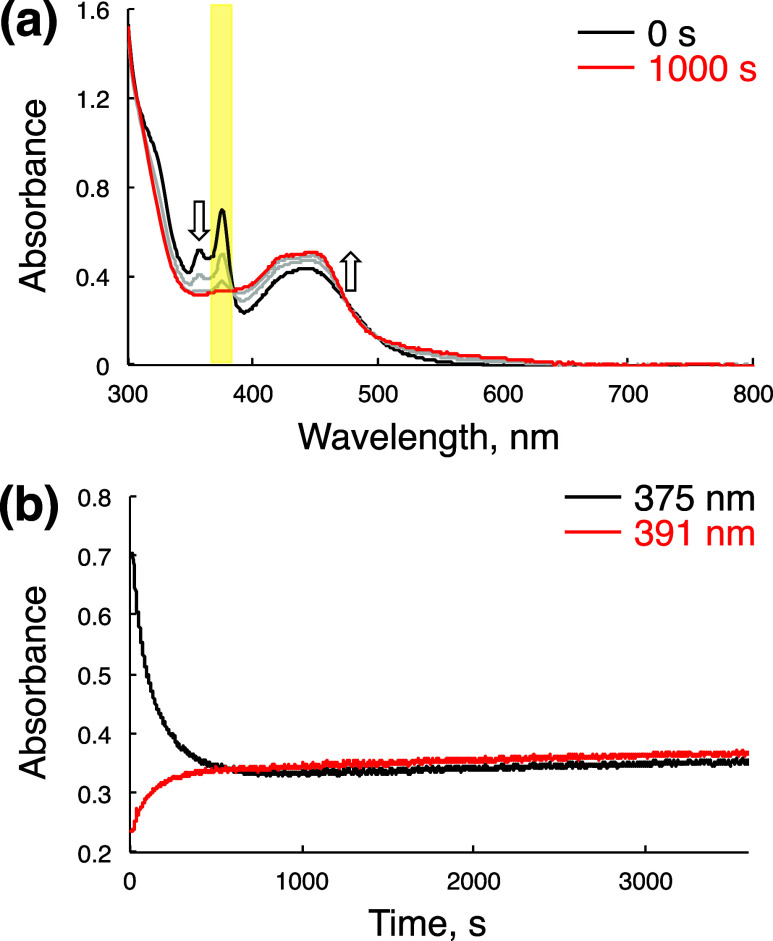
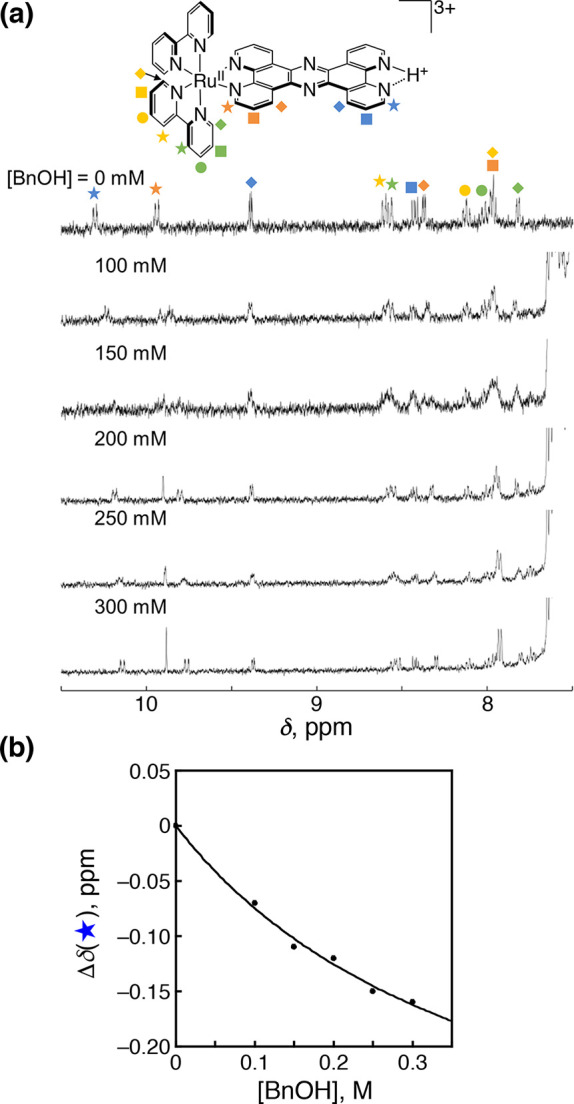
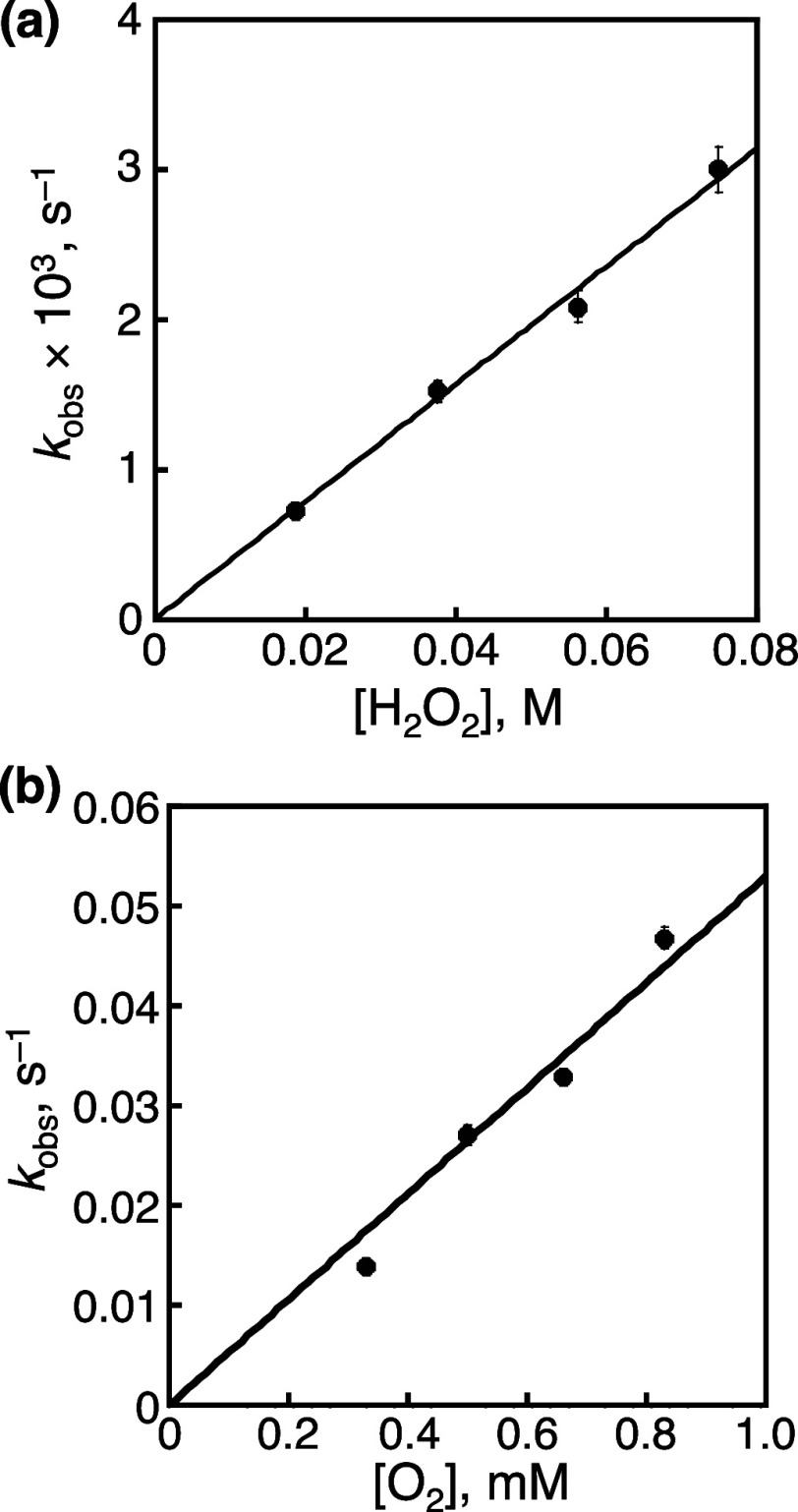
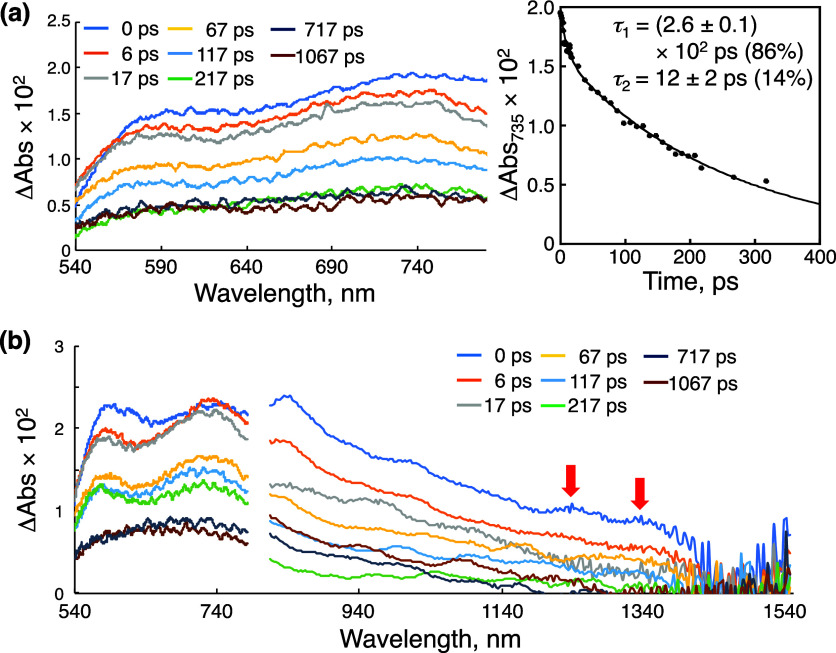
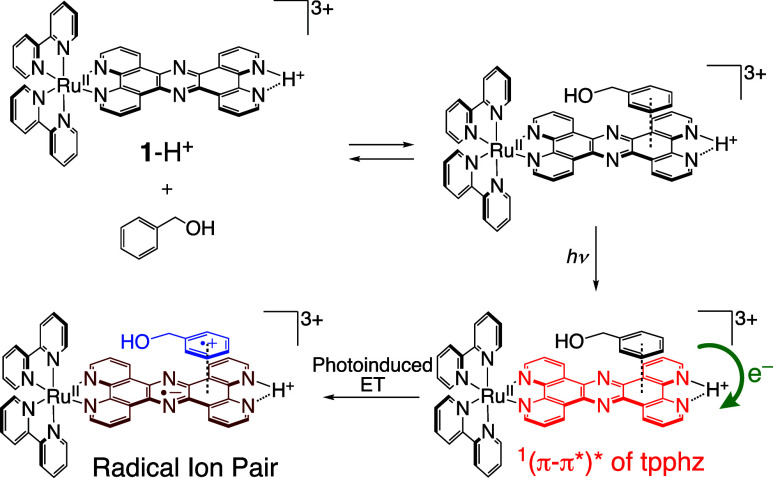
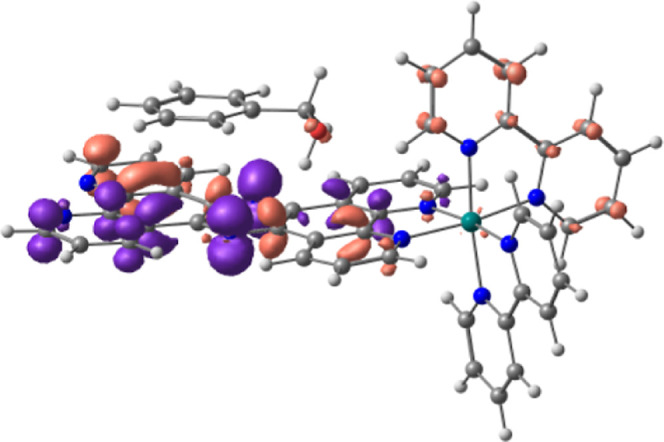
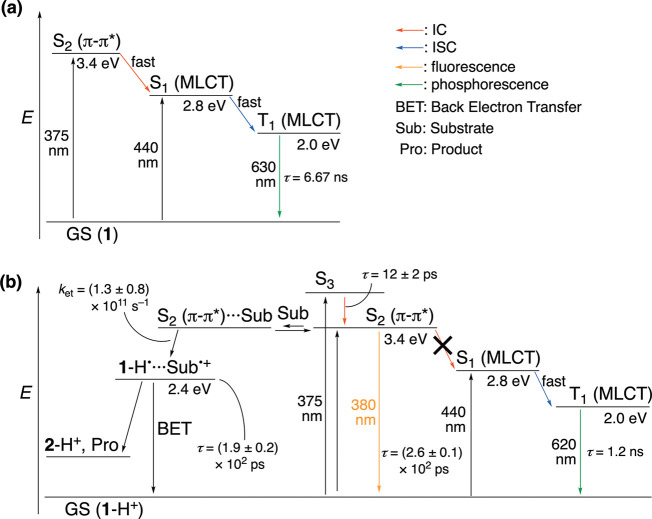
We have developed photocatalytic oxidation of aromatic substrates using O as a terminal oxidant to afford only 2e-oxidized products without the reductive activation of O in acidic water under visible-light irradiation. A Ru complex () bearing a pyrazine moiety as the active site in tetrapyrido[3,2-:2',3'-:3″,2″-:2‴,3‴-]phenazine (tpphz) as a ligand was employed as a photocatalyst. The active species for the photocatalysis was revealed to be not complex itself but the protonated form, -H, protonated at the vacant diimine site of tpphz. Upon photoexcitation in the presence of an organic substrate, -H was converted to the corresponding dihydro-intermediate (-H), where the pyrazine moiety of the ligand received 2e and 2H from the substrate. -H was facilely oxidized by O to recover -H. Consequently, an oxidation product of the substrate and HO derived from dioxygen reduction were obtained; however, the HO formed was also used for oxidation of -H. In the oxidation of benzyl alcohol to benzaldehyde, the turnover number reached 240 for 10 h, and the quantum yield was determined to be 4.0%. The absence of ring-opening products in the oxidation of cyclobutanol suggests that the catalytic reaction proceeds through a mechanism involving formal hydride transfer. Mechanistic studies revealed that the photocatalytic substrate oxidation by -H was achieved in neither the lowest singlet excited state nor triplet excited state (S or T) but in the second lowest singlet excited state (S), i.e., (π-π*)* of the tpphz ligand. Thus, the photocatalytic substrate oxidation by -H can be categorized into unusual anti-Kasha photocatalysis.
我们已经开发出一种利用氧气作为终端氧化剂对芳香族底物进行光催化氧化的方法,在可见光照射下的酸性水溶液中,该方法仅生成经过双电子氧化的产物,且不会使氧气发生还原活化。一种以四吡啶并[3,2 - a:2',3'- b:3″,2″- c:2‴,3‴- e]吩嗪(tpphz)为配体、带有吡嗪部分作为活性位点的钌配合物()被用作光催化剂。研究表明,光催化的活性物种并非配合物本身,而是在tpphz的空二亚胺位点质子化形成的质子化形式,即 -H。在有机底物存在下进行光激发时,-H会转化为相应的二氢中间体(-H),此时配体的吡嗪部分从底物接收2个电子和2个氢原子。-H很容易被氧气氧化以恢复为 -H。因此,得到了底物的氧化产物和由双氧还原产生的过氧化氢;然而,生成的过氧化氢也用于 -H的氧化。在将苯甲醇氧化为苯甲醛的反应中,10小时内的周转数达到240,量子产率测定为4.0%。环丁醇氧化过程中没有开环产物,这表明催化反应是通过涉及形式上氢化物转移的机制进行的。机理研究表明,-H对底物的光催化氧化既不是在最低单重激发态也不是在三重激发态(S或T)下实现的,而是在第二低单重激发态(S),即tpphz配体的(π-π*) *态下实现的。因此,-H对底物的光催化氧化可归类为不寻常的反卡莎光催化。