College of Information Science and Technology, University of Nebraska at Omaha, Omaha, NE, 68182, USA.
BMC Microbiol. 2024 Nov 21;24(Suppl 1):490. doi: 10.1186/s12866-024-03633-6.
Advances in metagenome sequencing data continue to enable new methods for analyzing biological systems. When handling microbial profile data, metagenome sequencing has proven to be far more comprehensive than traditional methods such as 16s rRNA data, which rely on partial sequences. Microbial community profiling can be used to obtain key biological insights that pave the way for more accurate understanding of complex systems that are critical for advancing biomedical research and healthcare. However, such attempts have mostly used partial or incomplete data to accurately capture those associations.
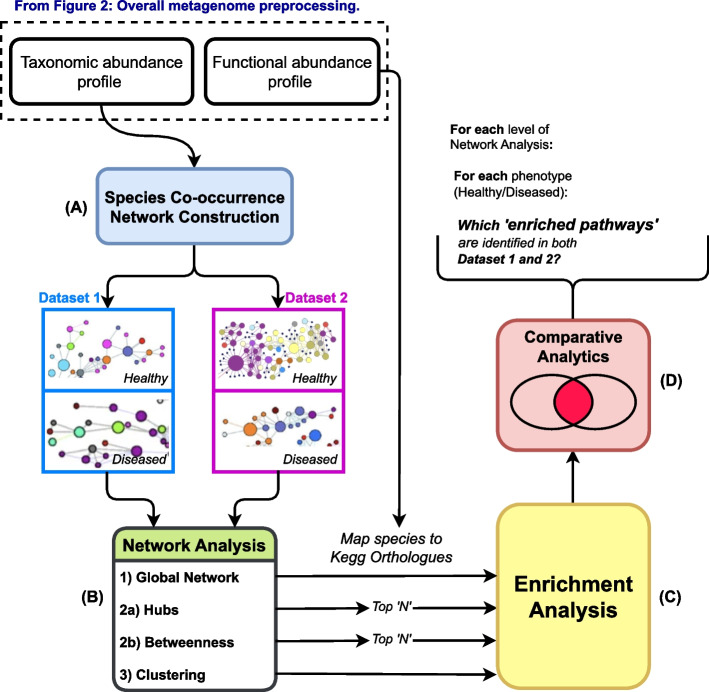
This study introduces a novel computational approach for the identification of co-occurring microbial communities using the abundance and functional roles of species-level microbiome data. The proposed approach is then used to identify signature pathways associated with inflammatory bowel disease (IBD). Furthermore, we developed a computational pipeline to identify microbial species co-occurrences from metagenome data at various granularity levels.
When comparing the IBD group to a control group, we show that certain co-occurring communities of species are enriched for potential pathways. We also show that the identified co-occurring microbial species operate as a community to facilitate pathway enrichment.
The obtained findings suggest that the proposed network model, along with the computational pipeline, provide a valuable analytical tool to analyze complex biological systems and extract pathway signatures that can be used to diagnose certain health conditions.
宏基因组测序数据的进步不断为分析生物系统的新方法提供支持。在处理微生物分布数据时,宏基因组测序已被证明远比传统方法如依赖于部分序列的 16s rRNA 数据全面。微生物群落分布分析可用于获得关键的生物学见解,为更准确地理解对推进生物医学研究和医疗保健至关重要的复杂系统铺平道路。然而,此类尝试大多使用部分或不完整的数据来准确捕捉这些关联。
本研究提出了一种新的计算方法,用于使用物种水平微生物组数据的丰度和功能角色识别共同出现的微生物群落。然后,我们使用该方法识别与炎症性肠病(IBD)相关的特征途径。此外,我们开发了一种计算管道,用于从不同粒度级别的宏基因组数据中识别微生物物种共现。
在将 IBD 组与对照组进行比较时,我们表明某些物种的共现群落富含潜在途径。我们还表明,鉴定出的共现微生物物种作为一个群落起作用,以促进途径富集。
获得的研究结果表明,所提出的网络模型和计算管道为分析复杂生物系统和提取可用于诊断某些健康状况的途径特征提供了有价值的分析工具。