Department of Information Engineering, University of Padova, Padova, Italy.
Department of Comparative Biomedicine and Food Science, University of Padova, Padova, Italy.
PLoS Comput Biol. 2022 Sep 8;18(9):e1010467. doi: 10.1371/journal.pcbi.1010467. eCollection 2022 Sep.
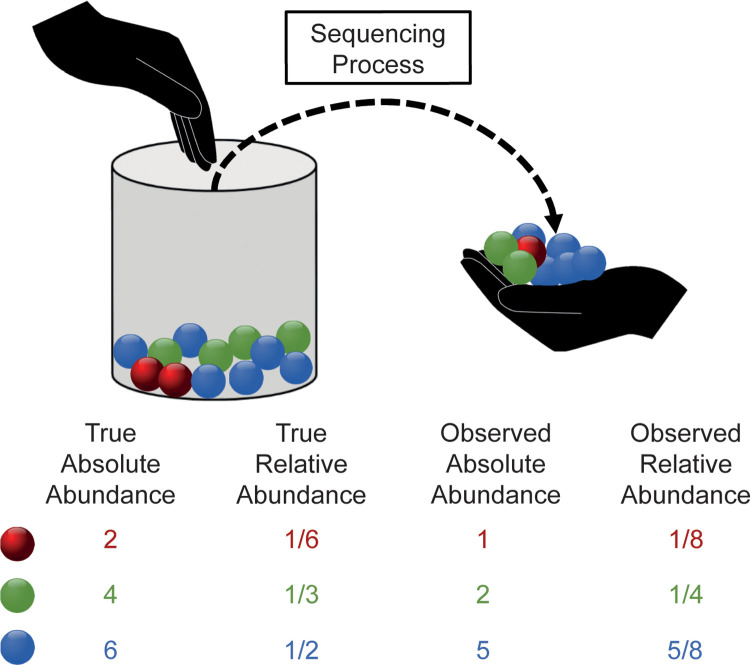
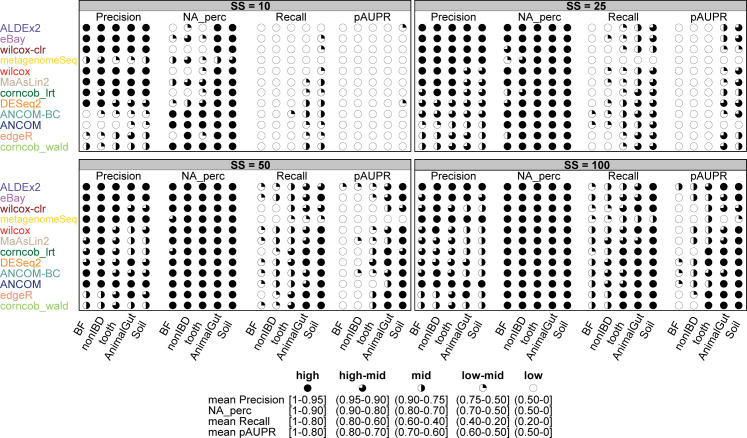
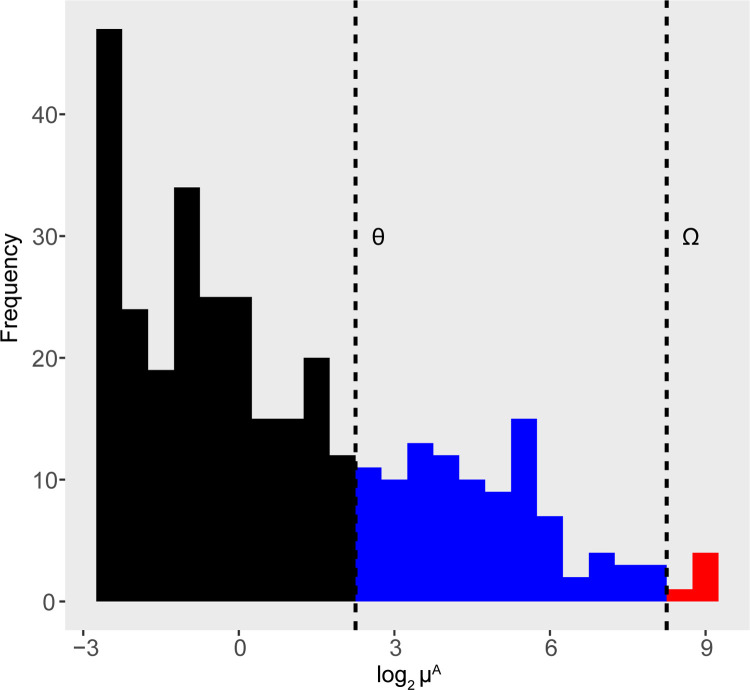
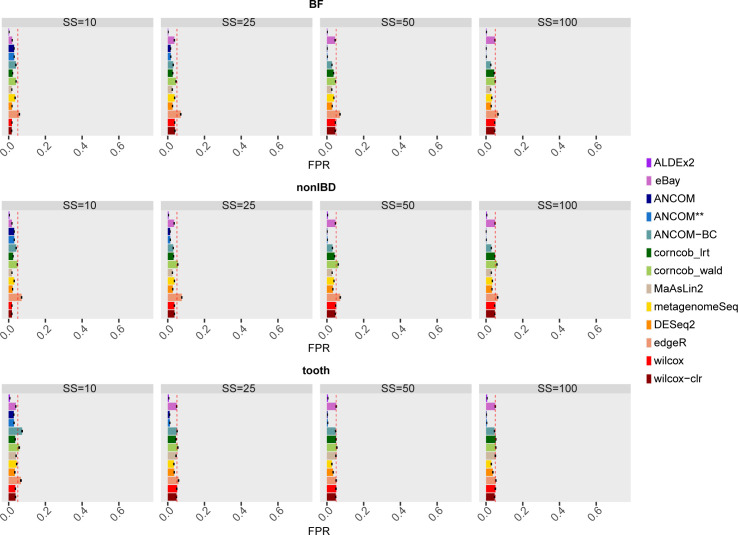
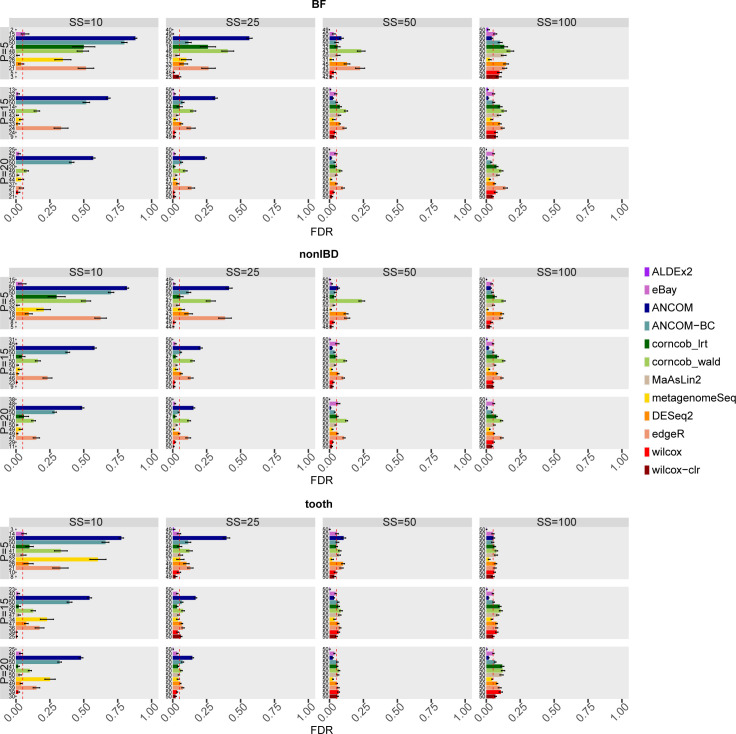
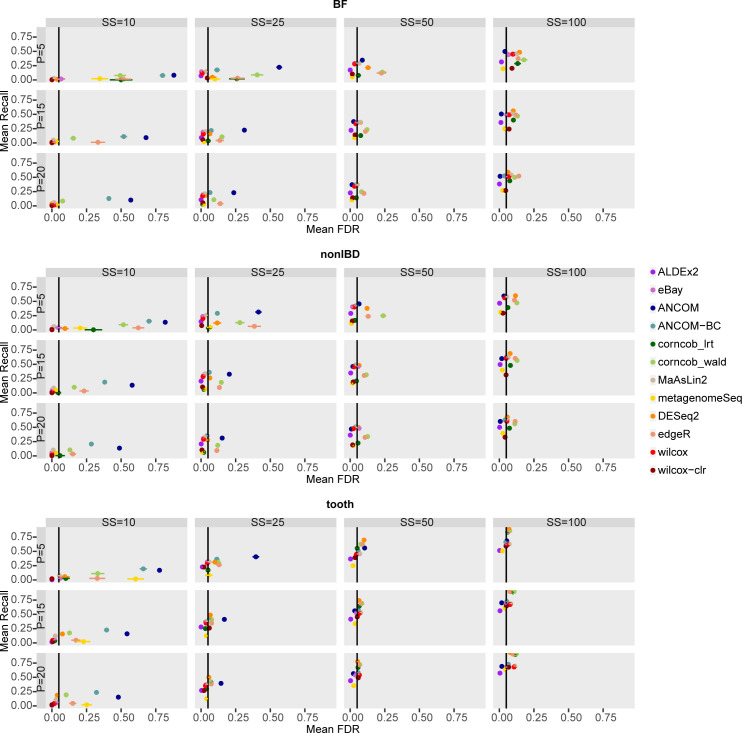
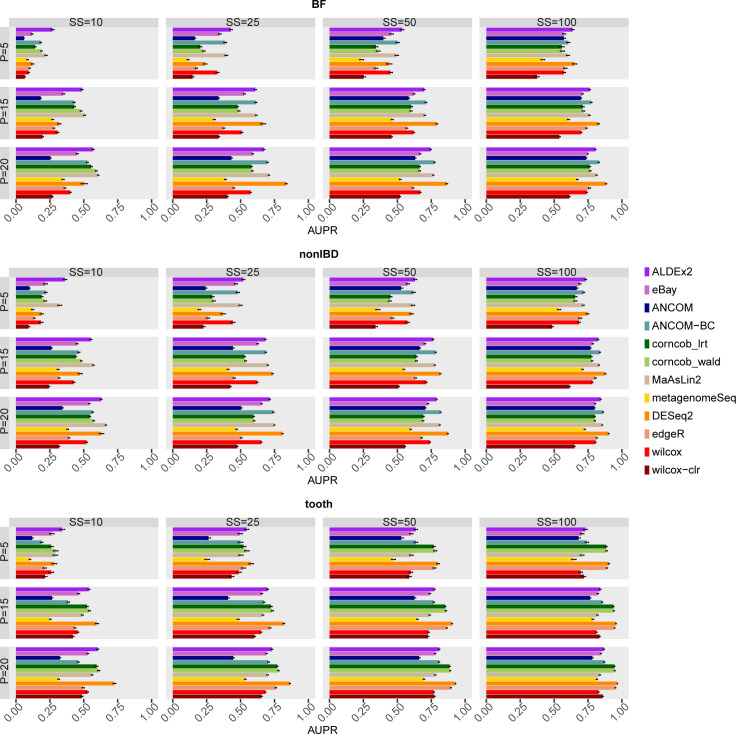
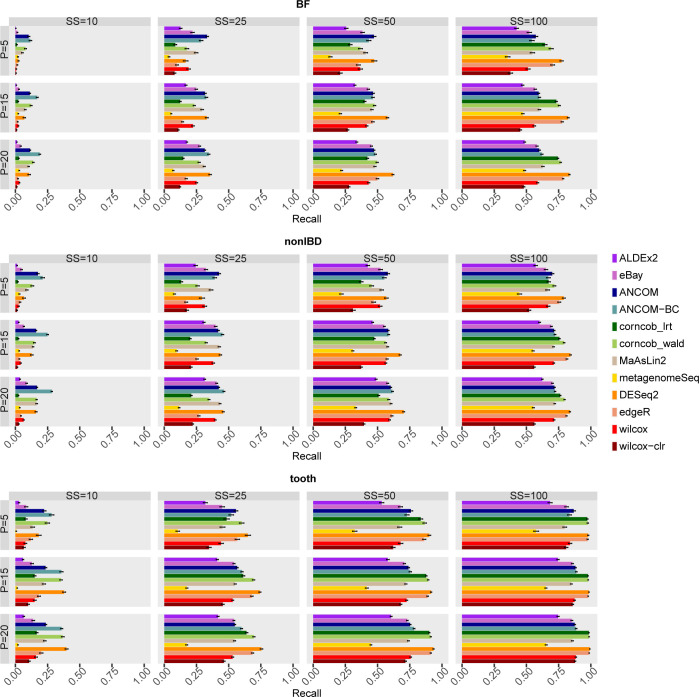
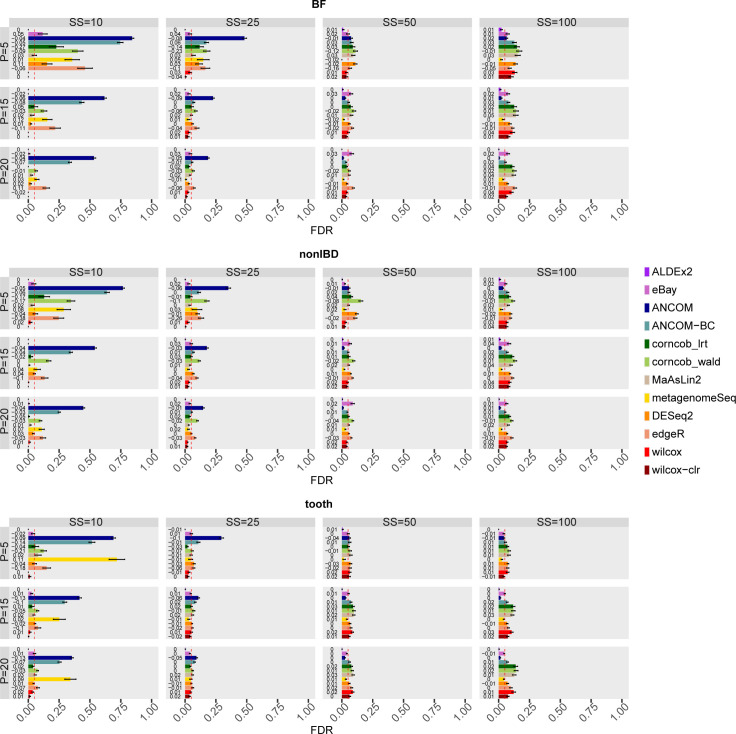
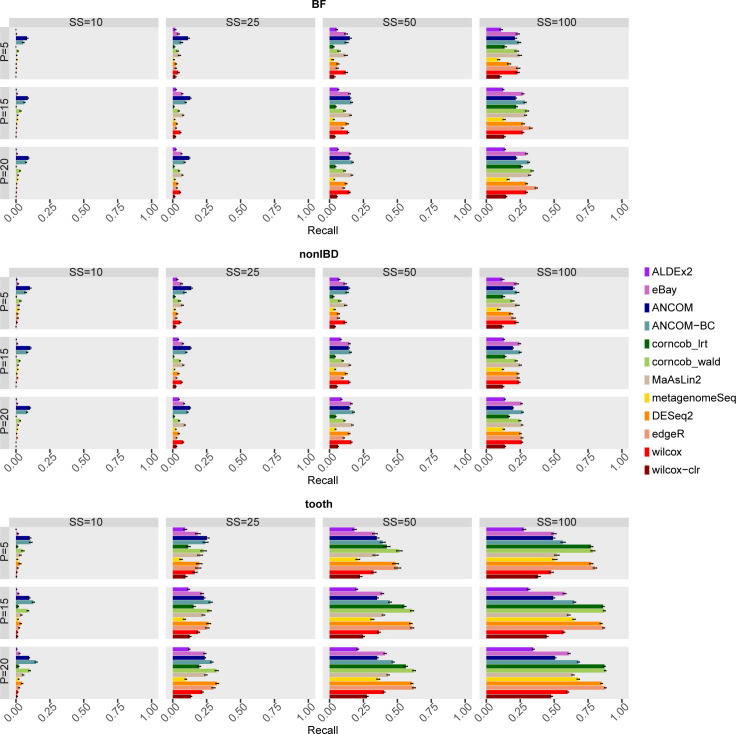
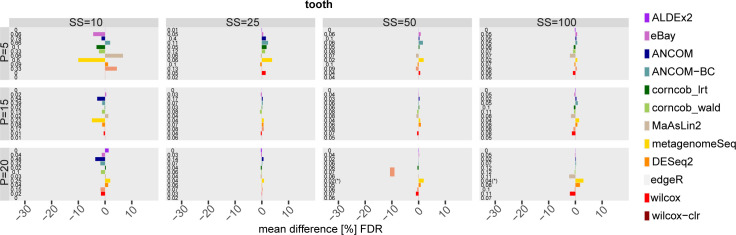
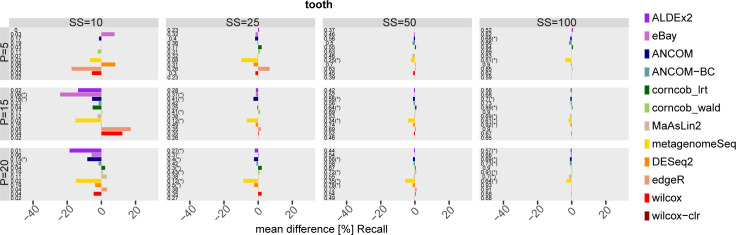
The development of increasingly efficient and cost-effective high throughput DNA sequencing techniques has enhanced the possibility of studying complex microbial systems. Recently, researchers have shown great interest in studying the microorganisms that characterise different ecological niches. Differential abundance analysis aims to find the differences in the abundance of each taxa between two classes of subjects or samples, assigning a significance value to each comparison. Several bioinformatic methods have been specifically developed, taking into account the challenges of microbiome data, such as sparsity, the different sequencing depth constraint between samples and compositionality. Differential abundance analysis has led to important conclusions in different fields, from health to the environment. However, the lack of a known biological truth makes it difficult to validate the results obtained. In this work we exploit metaSPARSim, a microbial sequencing count data simulator, to simulate data with differential abundance features between experimental groups. We perform a complete comparison of recently developed and established methods on a common benchmark with great effort to the reliability of both the simulated scenarios and the evaluation metrics. The performance overview includes the investigation of numerous scenarios, studying the effect on methods' results on the main covariates such as sample size, percentage of differentially abundant features, sequencing depth, feature variability, normalisation approach and ecological niches. Mainly, we find that methods show a good control of the type I error and, generally, also of the false discovery rate at high sample size, while recall seem to depend on the dataset and sample size.
高通量 DNA 测序技术的不断发展和成本效益的提高,增加了研究复杂微生物系统的可能性。最近,研究人员对研究不同生态位特征的微生物表现出了极大的兴趣。差异丰度分析旨在发现两组受试者或样本之间每个分类群丰度的差异,并为每个比较分配一个显著值。已经专门开发了几种生物信息学方法,考虑到微生物组数据的挑战,例如稀疏性、样本之间不同测序深度的约束和组成性。差异丰度分析在从健康到环境的不同领域得出了重要结论。然而,缺乏已知的生物学事实使得难以验证所获得的结果。在这项工作中,我们利用 metaSPARSim,一种微生物测序计数数据模拟器,在实验分组之间具有差异丰度特征的模拟数据。我们在一个共同的基准上对最近开发和建立的方法进行了全面的比较,非常注重模拟场景和评估指标的可靠性。性能概述包括对许多场景的研究,研究主要协变量(例如样本量、差异丰度特征的百分比、测序深度、特征可变性、归一化方法和生态位)对方法结果的影响。主要发现是,方法在高样本量时,对第一类错误和假发现率的控制较好,而召回率似乎取决于数据集和样本量。