AI Lab, Tencent, Shenzhen, China.
Department of Computer Science, Hunan University, Hunan, China.
Nat Commun. 2024 Nov 25;15(1):10223. doi: 10.1038/s41467-024-54440-6.
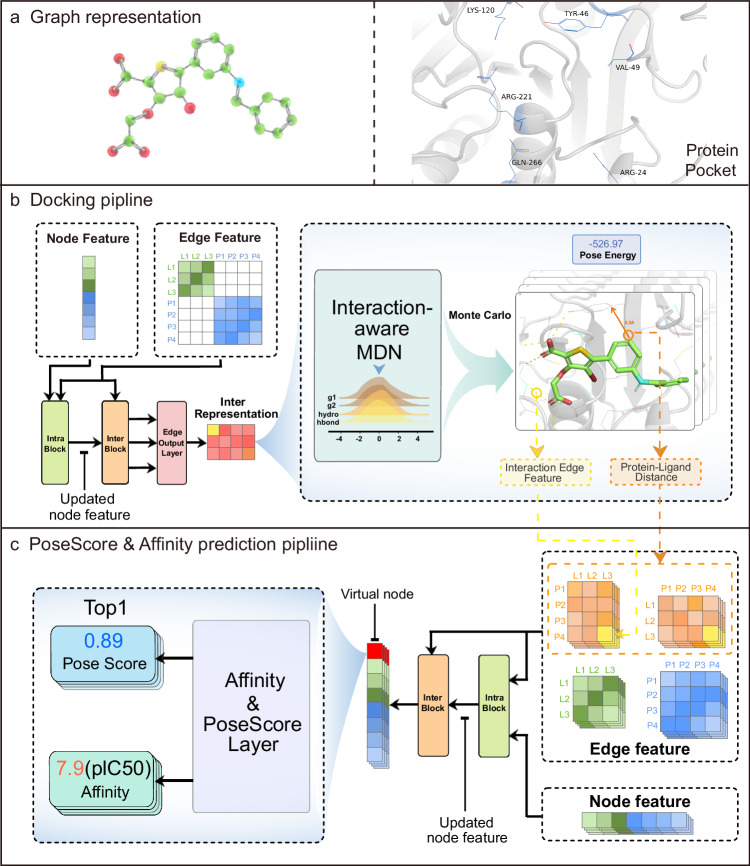
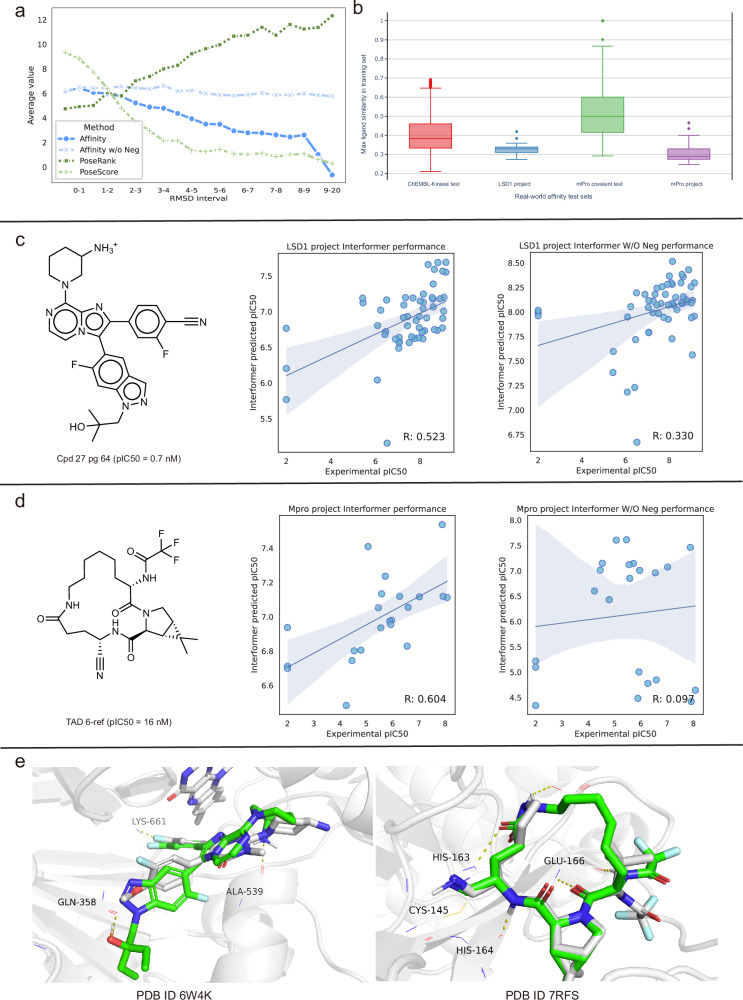
In recent years, the application of deep learning models to protein-ligand docking and affinity prediction, both vital for structure-based drug design, has garnered increasing interest. However, many of these models overlook the intricate modeling of interactions between ligand and protein atoms in the complex, consequently limiting their capacity for generalization and interpretability. In this work, we propose Interformer, a unified model built upon the Graph-Transformer architecture. The proposed model is designed to capture non-covalent interactions utilizing an interaction-aware mixture density network. Additionally, we introduce a negative sampling strategy, facilitating an effective correction of interaction distribution for affinity prediction. Experimental results on widely used and our in-house datasets demonstrate the effectiveness and universality of the proposed approach. Extensive analyses confirm our claim that our approach improves performance by accurately modeling specific protein-ligand interactions. Encouragingly, our approach advances docking tasks state-of-the-art (SOTA) performance.
近年来,深度学习模型在蛋白质配体对接和亲和力预测中的应用引起了越来越多的关注,这两个方面对于基于结构的药物设计至关重要。然而,这些模型中的许多模型忽略了复合物中配体和蛋白质原子之间相互作用的复杂建模,从而限制了它们的泛化能力和可解释性。在这项工作中,我们提出了 Interformer,这是一个基于图变换架构的统一模型。所提出的模型旨在利用交互感知混合密度网络来捕获非共价相互作用。此外,我们引入了一种负采样策略,有助于有效地纠正亲和力预测中的相互作用分布。在广泛使用的数据集和我们内部数据集上的实验结果表明了所提出方法的有效性和通用性。广泛的分析证实了我们的观点,即我们的方法通过准确地建模特定的蛋白质-配体相互作用来提高性能。令人鼓舞的是,我们的方法提高了对接任务的 SOTA 性能。