Department of Bio and Brain Engineering, Korea Advanced Institute of Science and Technology, Daejeon, South Korea.
PLoS One. 2021 Apr 8;16(4):e0249404. doi: 10.1371/journal.pone.0249404. eCollection 2021.
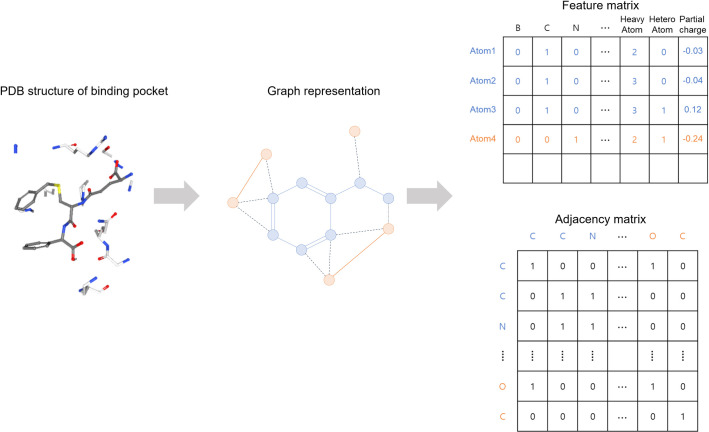
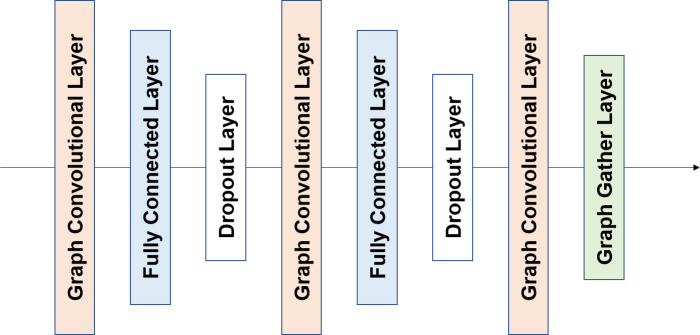
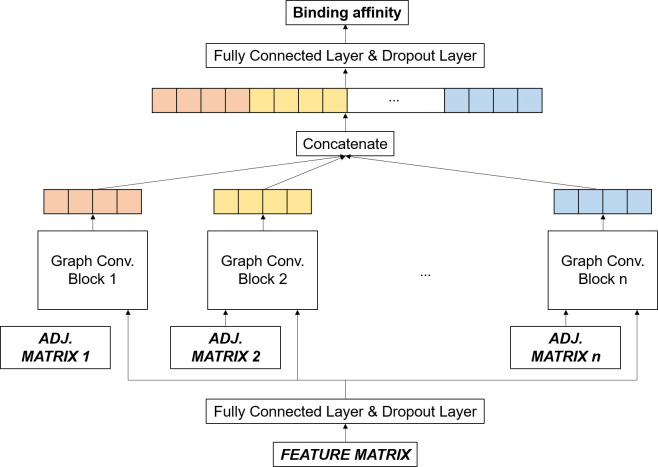
Prediction of protein-ligand interactions is a critical step during the initial phase of drug discovery. We propose a novel deep-learning-based prediction model based on a graph convolutional neural network, named GraphBAR, for protein-ligand binding affinity. Graph convolutional neural networks reduce the computational time and resources that are normally required by the traditional convolutional neural network models. In this technique, the structure of a protein-ligand complex is represented as a graph of multiple adjacency matrices whose entries are affected by distances, and a feature matrix that describes the molecular properties of the atoms. We evaluated the predictive power of GraphBAR for protein-ligand binding affinities by using PDBbind datasets and proved the efficiency of the graph convolution. Given the computational efficiency of graph convolutional neural networks, we also performed data augmentation to improve the model performance. We found that data augmentation with docking simulation data could improve the prediction accuracy although the improvement seems not to be significant. The high prediction performance and speed of GraphBAR suggest that such networks can serve as valuable tools in drug discovery.
蛋白质-配体相互作用的预测是药物发现初始阶段的关键步骤。我们提出了一种基于图卷积神经网络的新型深度学习预测模型,称为 GraphBAR,用于预测蛋白质-配体结合亲和力。图卷积神经网络减少了传统卷积神经网络模型通常所需的计算时间和资源。在该技术中,蛋白质-配体复合物的结构表示为多个邻接矩阵的图,其条目受距离影响,以及描述原子分子特性的特征矩阵。我们使用 PDBbind 数据集评估了 GraphBAR 对蛋白质-配体结合亲和力的预测能力,并证明了图卷积的有效性。鉴于图卷积神经网络的计算效率,我们还进行了数据扩充以提高模型性能。我们发现,使用对接模拟数据进行数据扩充可以提高预测准确性,尽管这种改进似乎并不显著。GraphBAR 的高预测性能和速度表明,此类网络可以成为药物发现的有价值工具。