Jardanowska-Kotuniak Marta, Dramiński Michał, Własnowolski Michał, Łapiński Marcin, Sengupta Kaustav, Agarwal Abhishek, Filip Adam, Ghosh Nimisha, Pancaldi Vera, Grynberg Marcin, Saha Indrajit, Plewczynski Dariusz, Dąbrowski Michał J
Computational Biology Group, Institute of Computer Science of the Polish Academy of Sciences, Warsaw, Poland.
Institute of Biochemistry and Biophysics of the Polish Academy of Sciences, Warsaw, Poland.
bioRxiv. 2024 Nov 15:2024.11.12.623187. doi: 10.1101/2024.11.12.623187.
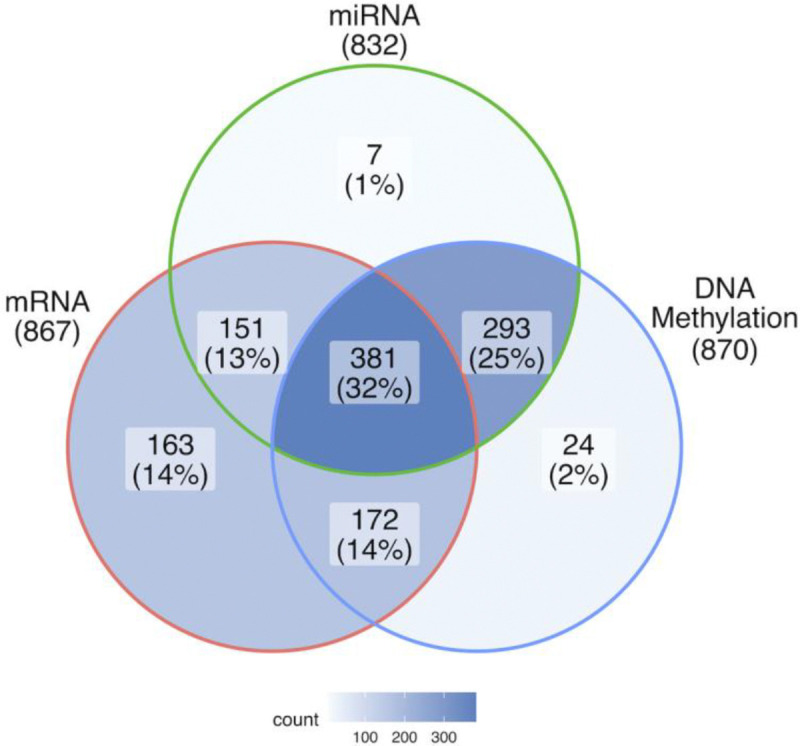
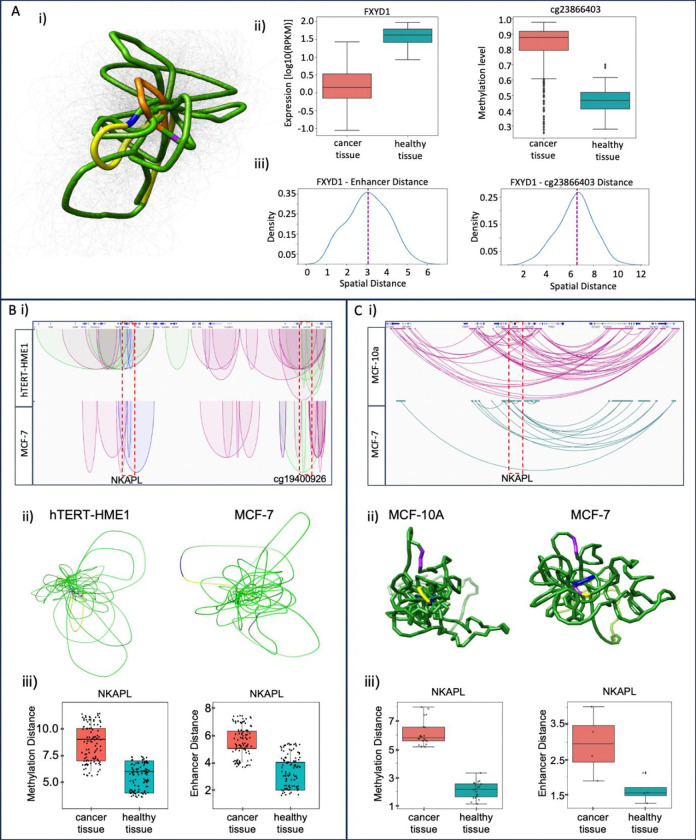
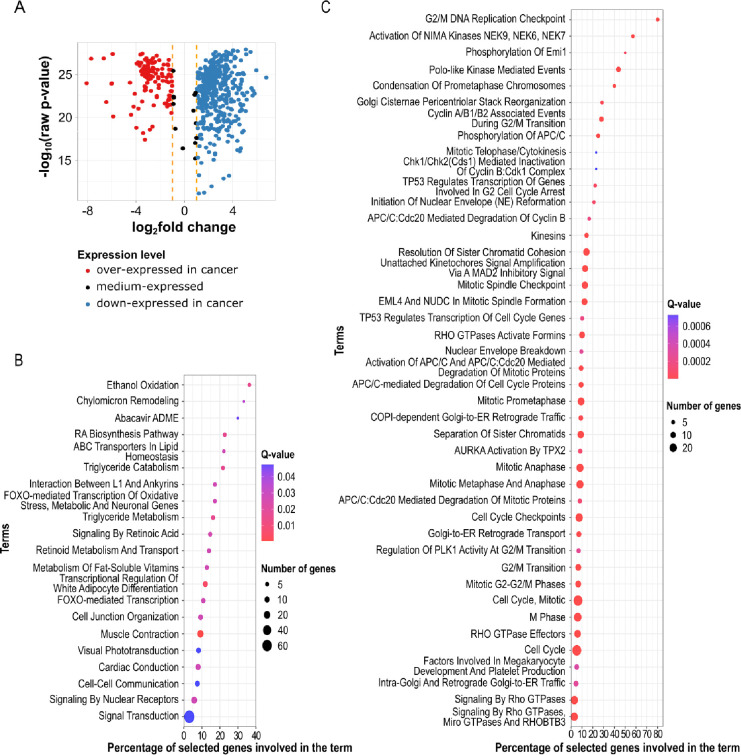
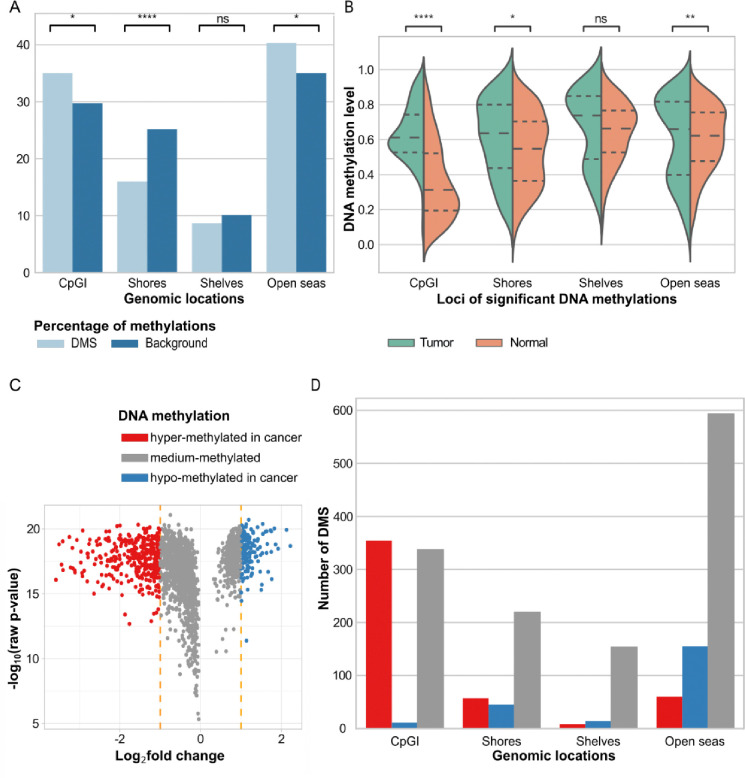
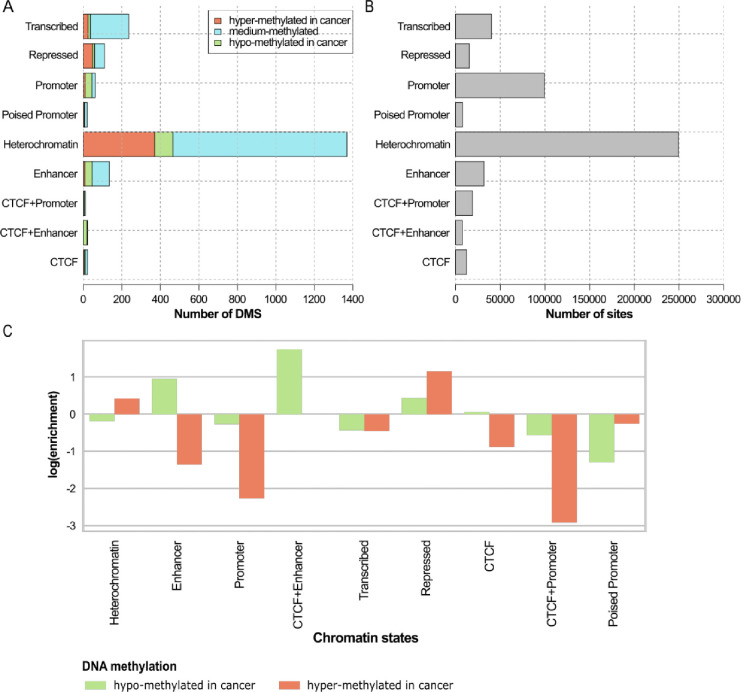
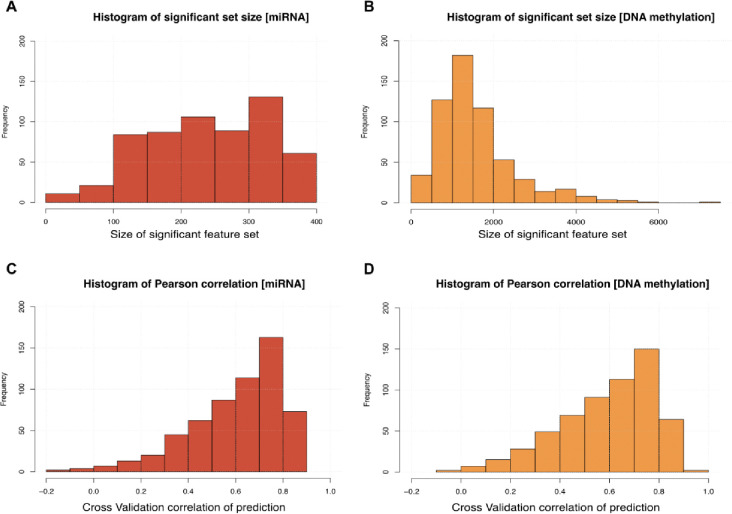
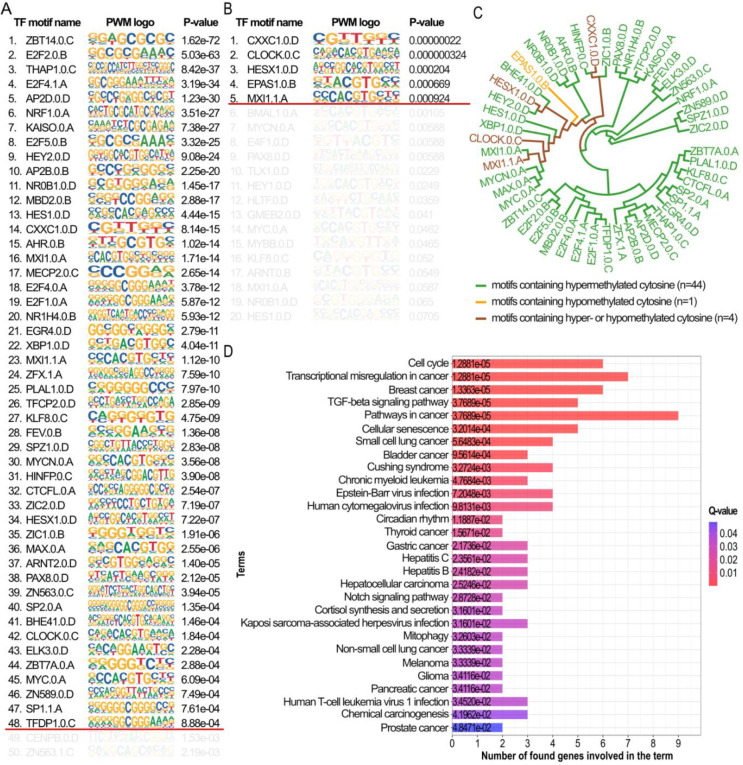
Breast cancer is the most common cancer in women and the 2nd most common cancer worldwide, yearly impacting over 2 million females and causing 650 thousand deaths. It has been widely studied, but its epigenetic variation is not entirely unveiled. We aimed to identify epigenetic mechanisms impacting the expression of breast cancer related genes to detect new potential biomarkers and therapeutic targets. We considered The Cancer Genome Atlas database with over 800 samples and several omics datasets such as mRNA, miRNA, DNA methylation, which we used to select 2701 features that were statistically significant to differ between cancer and control samples using the Monte Carlo Feature Selection and Interdependency Discovery algorithm, from an initial total of 417,486. Their biological impact on cancerogenesis was confirmed using: statistical analysis, natural language processing, linear and machine learning models as well as: transcription factors identification, drugs and 3D chromatin structure analyses. Classification of cancer vs control samples on the selected features returned high classification weighted Accuracy from 0.91 to 0.98 depending on feature-type: mRNA, miRNA, DNA methylation, and classification algorithm. In general, cancer samples showed lower expression of differentially expressed genes and increased -values of differentially methylated sites. We identified mRNAs whose expression is well explained by miRNA expression and differentially methylated sites -values. We recognized differentially methylated sites possibly affecting NRF1 and MXI1 transcription factors binding, causing a disturbance in and expression, respectively. Our 3D models showed more loosely packed chromatin in cancer. This study successfully points out numerous possible regulatory dependencies.
乳腺癌是女性中最常见的癌症,也是全球第二常见的癌症,每年影响超过200万女性并导致65万人死亡。虽然它已得到广泛研究,但其表观遗传变异尚未完全揭示。我们旨在确定影响乳腺癌相关基因表达的表观遗传机制,以检测新的潜在生物标志物和治疗靶点。我们考虑了拥有800多个样本的癌症基因组图谱数据库以及几个组学数据集,如mRNA、miRNA、DNA甲基化,我们使用蒙特卡罗特征选择和相关性发现算法从最初总共417486个特征中选择了2701个在癌症样本和对照样本之间具有统计学显著差异的特征。使用统计分析、自然语言处理、线性和机器学习模型,以及转录因子鉴定、药物和3D染色质结构分析,证实了它们对肿瘤发生的生物学影响。根据特征类型(mRNA、miRNA、DNA甲基化)和分类算法,对选定特征进行癌症样本与对照样本的分类,返回的分类加权准确率较高,从0.91到0.98不等。一般来说,癌症样本中差异表达基因的表达较低,差异甲基化位点的 值增加。我们鉴定出其表达可由miRNA表达和差异甲基化位点 值很好解释的mRNA。我们识别出可能影响NRF1和MXI1转录因子结合的差异甲基化位点,分别导致 和 表达紊乱。我们的3D模型显示癌症中染色质包装更松散。本研究成功指出了许多可能的调控依赖性。