School of Physiology, Pharmacology and Neuroscience, Biomedical Sciences Building, University of Bristol, University Walk, Bristol, BS8 1TD, UK.
Biomedical Imaging Research Centre, University of Fukui, 23-3 Matsuokashimoaizuki, Eiheiji-cho, Fukui, 910-1193, Japan.
Cell Mol Life Sci. 2024 Nov 28;81(1):466. doi: 10.1007/s00018-024-05498-4.
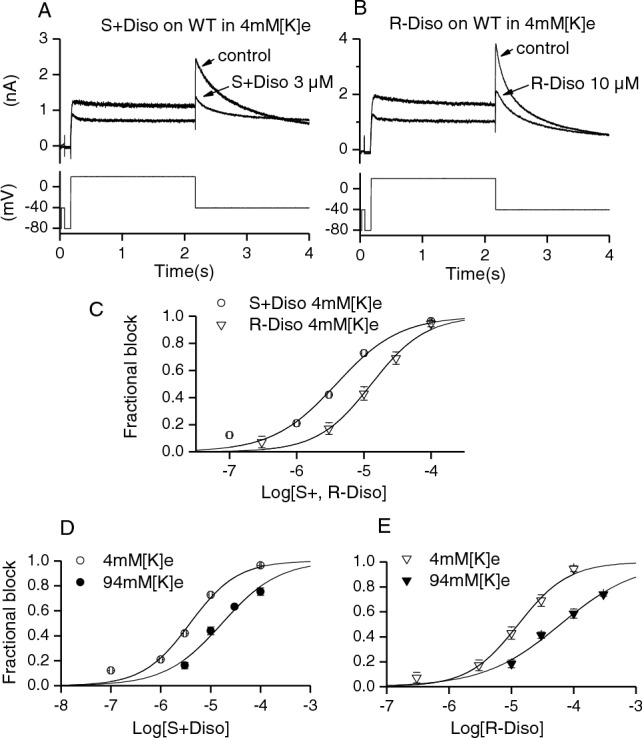
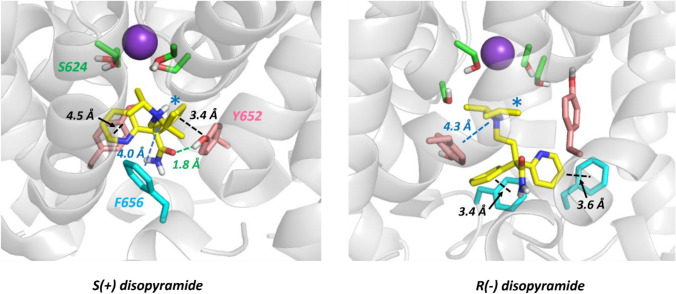
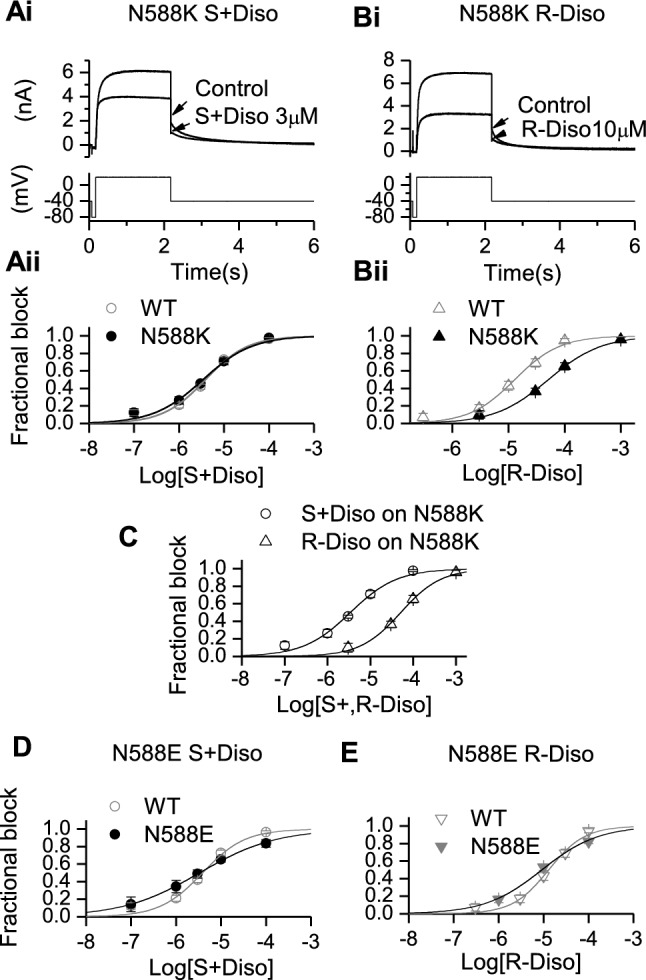
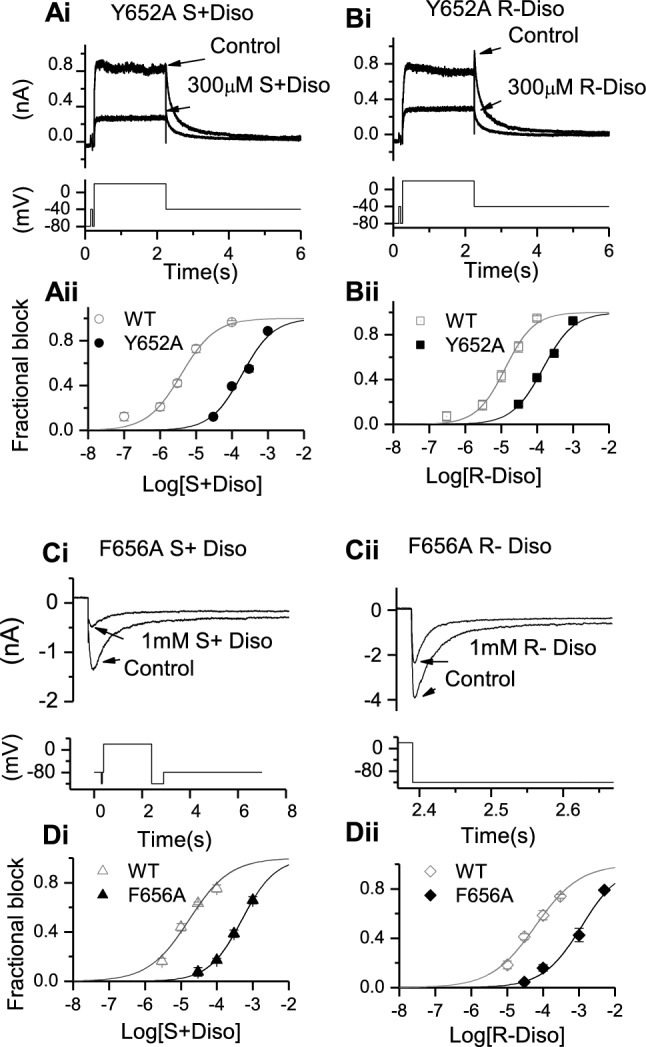
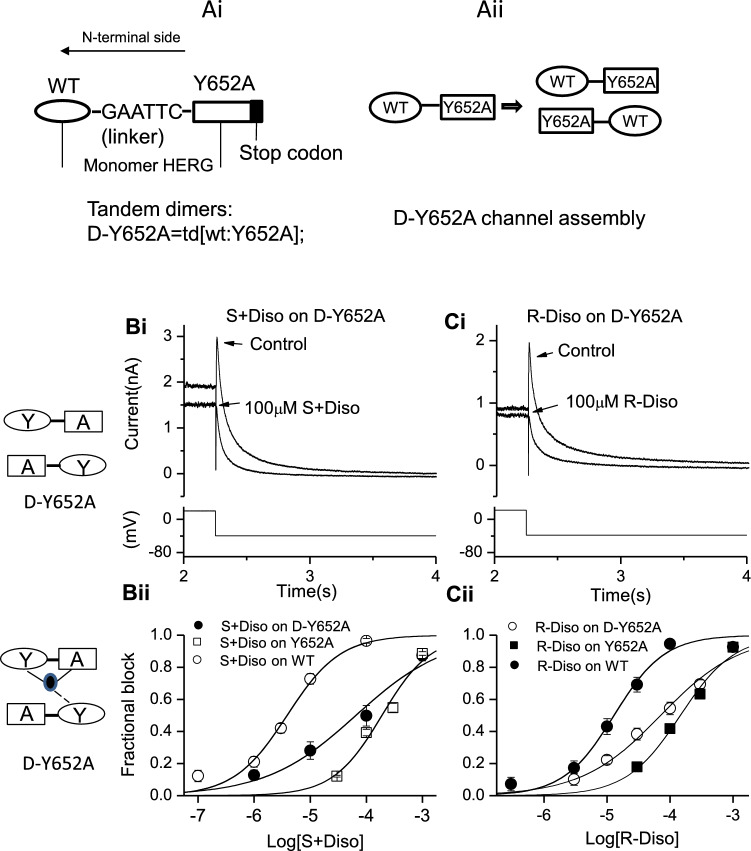
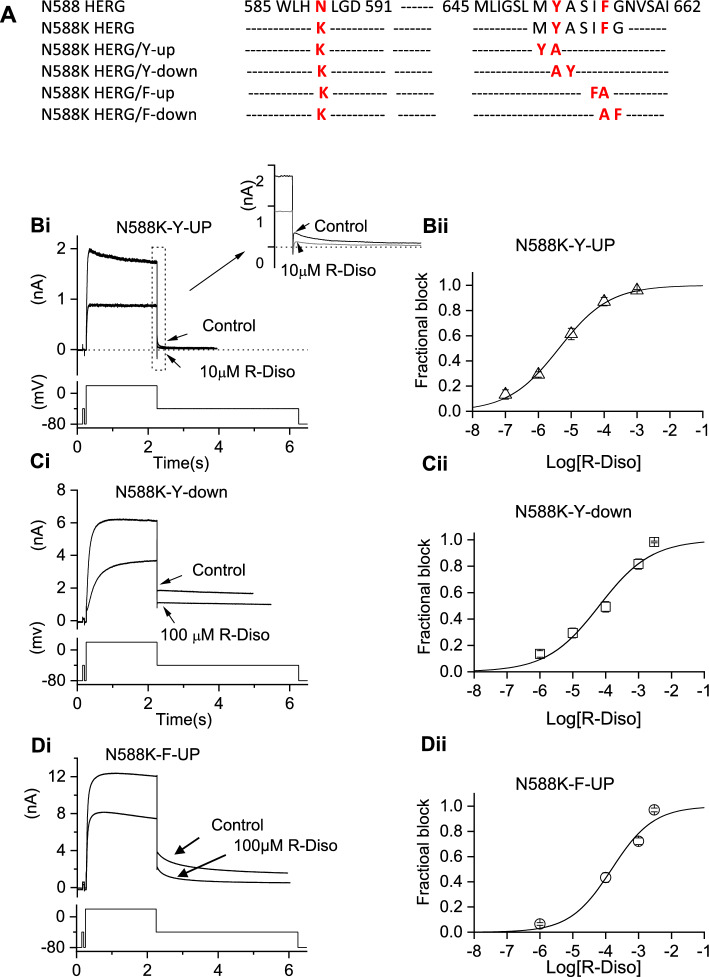
Potassium channels encoded by human Ether-à-go-go-Related Gene (hERG) are inhibited by diverse cardiac and non-cardiac drugs. Disopyramide is a chiral Class Ia antiarrhythmic that inhibits hERG at clinical concentrations. This study evaluated effects of disopyramide enantiomers on hERG current (I) from hERG expressing HEK 293 cells at 37 °C. S(+) and R(-) disopyramide inhibited wild-type (WT) I with IC values of 3.9 µM and 12.9 µM respectively. The attenuated-inactivation mutant N588K had little effect on the action of S(+) disopyramide but the IC for the R(-) enantiomer was ~ 15-fold that for S(+) disopyramide. The enhanced inactivation mutant N588E only slightly increased the potency of R(-) disopyramide. S6 mutation Y652A reduced S(+) disopyramide potency more than that of R(-) disopyramide (respective IC values ~ 49-fold and 11-fold their WT controls). The F656A mutation also exerted a stronger effect on S(+) than R(-) disopyramide, albeit with less IC elevation. A WT-Y652A tandem dimer exhibited a sensitivity to the enantiomers that was intermediate between that of WT and Y652A, suggesting Y652 groups on adjacent subunits contribute to the binding. Moving the Y (normally at site 652) one residue in the N- terminal (up) direction in N588K hERG markedly increased the blocking potency of R(-) disopyramide. Molecular dynamics simulations using a hERG pore model produced different binding modes for S(+) and R(-) disopyramide consistent with the experimental observations. In conclusion, S(+) disopyramide interacts more strongly with S6 aromatic binding residues on hERG than does R(-) disopyramide, whilst optimal binding of the latter is more reliant on intact inactivation.
人 Ether-à-go-go 相关基因 (hERG) 编码的钾通道被多种心脏和非心脏药物抑制。普罗帕酮是一种手性 Ia 类抗心律失常药物,在临床浓度下抑制 hERG。本研究评估了普罗帕酮对 37°C 时表达 hERG 的 HEK 293 细胞 hERG 电流 (I) 的对映异构体的影响。S(+)和 R(-)普罗帕酮对野生型 (WT) I 的抑制作用的 IC 值分别为 3.9 µM 和 12.9 µM。衰减失活突变体 N588K 对 S(+)普罗帕酮的作用影响不大,但 R(-)对映异构体的 IC 值是 S(+)普罗帕酮的约 15 倍。增强失活突变体 N588E 仅略微增加了 R(-)普罗帕酮的效力。S6 突变 Y652A 降低了 S(+)普罗帕酮的效力,比 R(-)普罗帕酮更为显著(各自的 IC 值分别为其 WT 对照的 49 倍和 11 倍)。F656A 突变对 S(+)普罗帕酮的作用也比 R(-)普罗帕酮更强,尽管 IC 值升高幅度较小。WT-Y652A 串联二聚体对两种对映异构体的敏感性介于 WT 和 Y652A 之间,表明相邻亚基上的 Y652 基团有助于结合。将 Y(通常位于 652 位)在 N 端(向上)方向上移动一个残基,在 N588K hERG 中,显著增加了 R(-)普罗帕酮的阻断效力。使用 hERG 孔模型进行的分子动力学模拟产生了 S(+)和 R(-)普罗帕酮的不同结合模式,与实验观察结果一致。总之,S(+)普罗帕酮与 hERG 的 S6 芳香族结合残基的相互作用比 R(-)普罗帕酮更强,而后者的最佳结合更依赖于完整的失活。