Thaweesapphithak Sermporn, Termteerapornpimol Kittipat, Wongsirisuwan Siriwong, Chantarangsu Soranun, Porntaveetus Thantrira
Department of Physiology, Center of Excellence in Genomics and Precision Dentistry, Faculty of Dentistry, Chulalongkorn University, Bangkok, 10330, Thailand.
Department of Oral Pathology, Faculty of Dentistry, Chulalongkorn University, Bangkok, 10330, Thailand.
J Transl Med. 2024 Dec 3;22(1):1099. doi: 10.1186/s12967-024-05904-2.
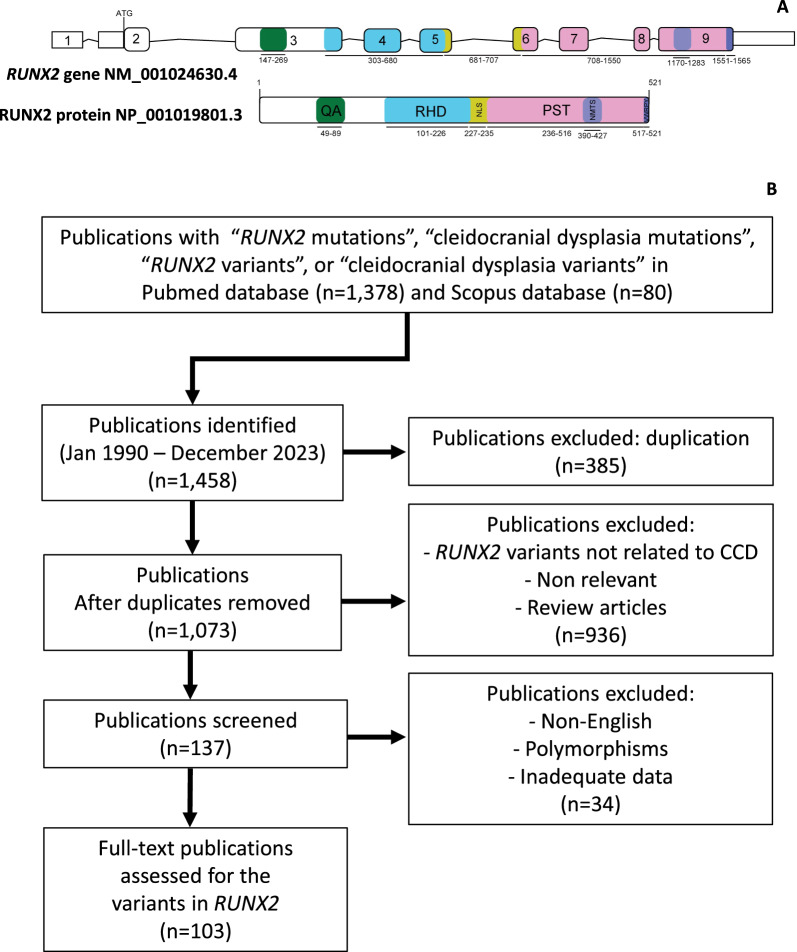
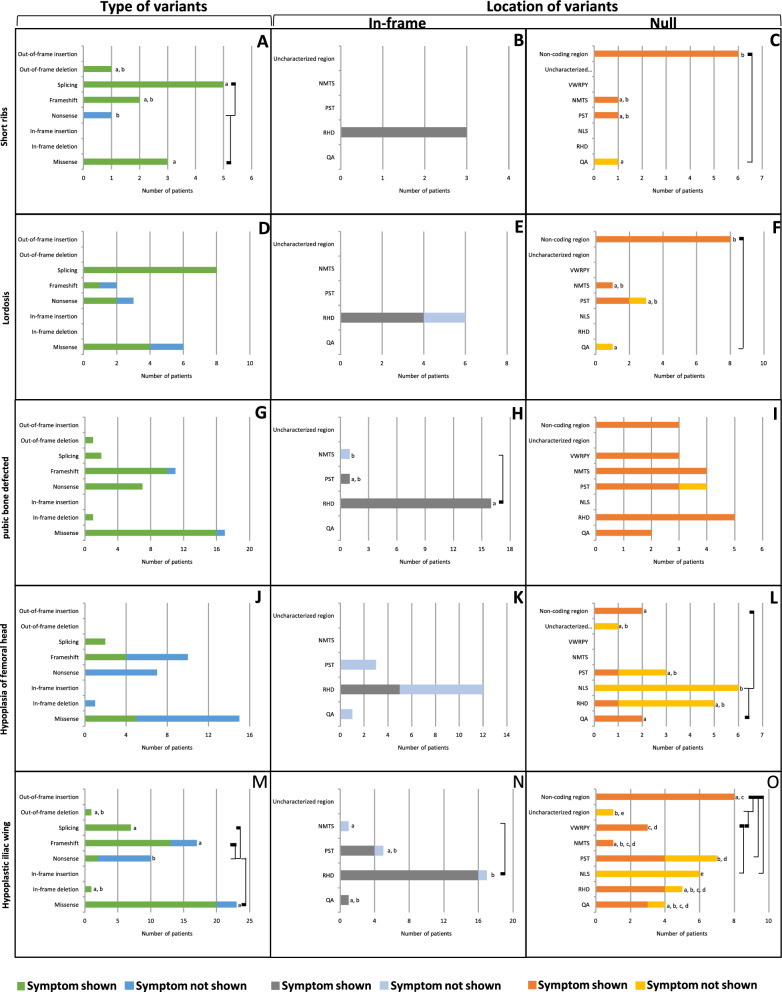
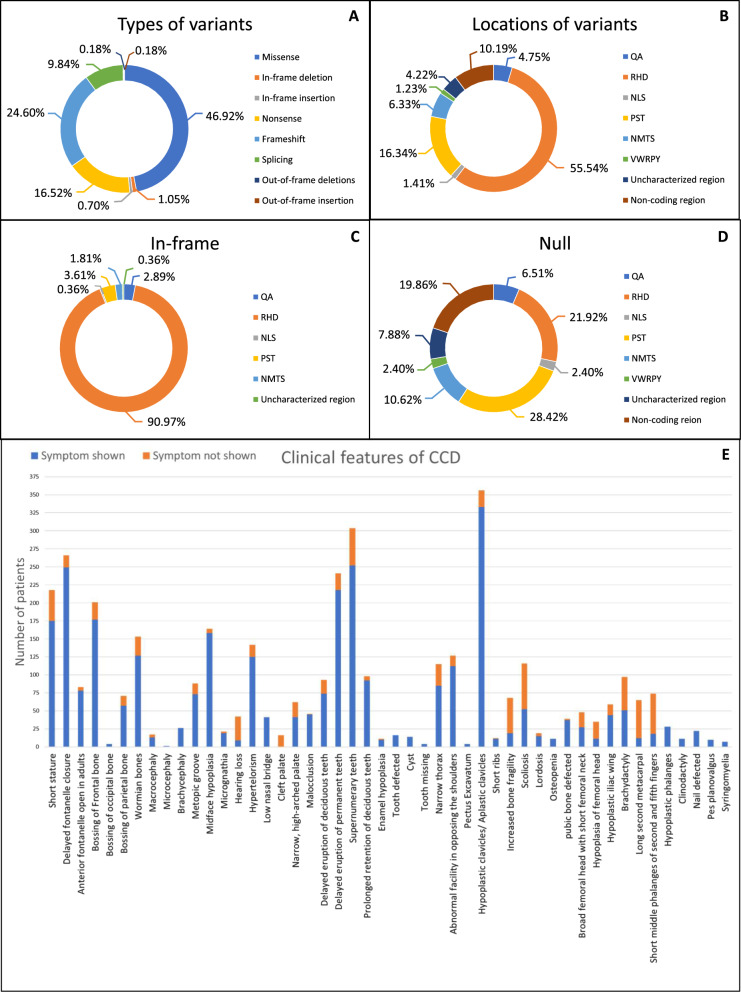
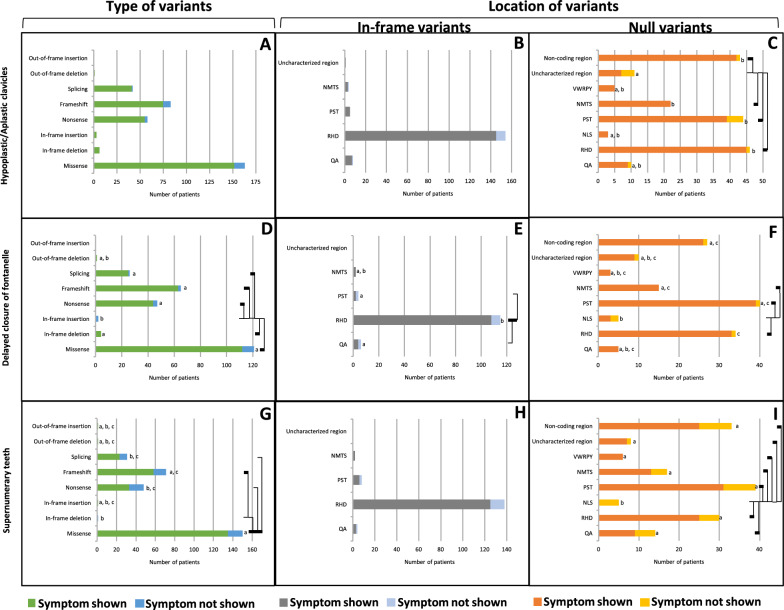
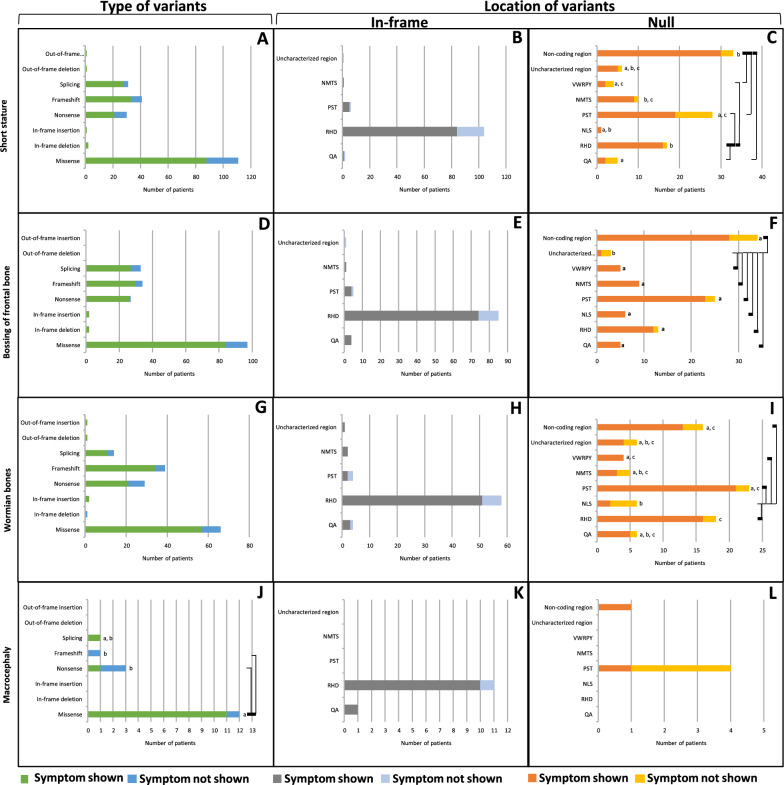
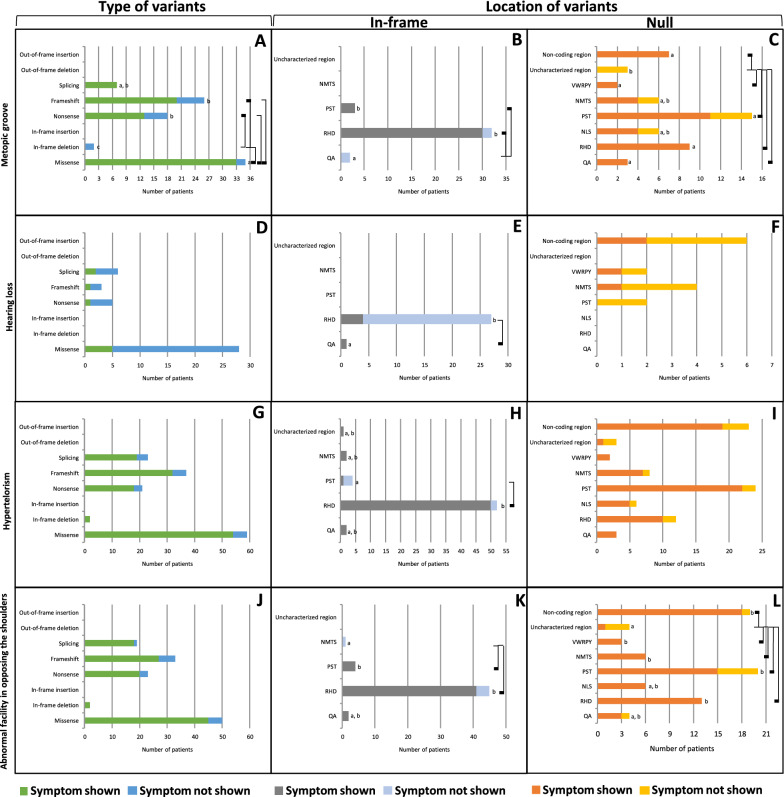
Cleidocranial Dysplasia (CCD) is a rare genetic disorder characterized by skeletal abnormalities and dental anomalies, primarily caused by variants in the RUNX2 gene. Understanding the spectrum of RUNX2 variants and their effects on CCD phenotypes is crucial for accurate diagnosis and management strategies. This systematic review aimed to comprehensively analyze the genotypic and phenotypic spectra of RUNX2 variants in CCD patients, assess their distribution across functional regions, and investigate genotype-phenotype correlations. This review included 569 reported variants and 453 CCD patients from 103 articles. Of 569 variants, in-frame variants constituted 48.68%, while null variants accounted for 51.32%. Regarding locations, RUNX2 variants were predominantly located in the RHD (55.54%), followed by PST (16.34%), NMTS (6.33%), QA (4.75%), VWRPY (1.23%), and NLS (1.41%) regions while 10.19% were in non-coding regions. In-frame variants occurred primarily in the RHD (90.97%), while null variants were found across various regions of RUNX2. Data analysis revealed a correlation between variant location and specific skeletal features in CCD patients. Missense variants, predominantly found within the functionally critical RHD, were significantly associated with supernumerary teeth, macrocephaly, metopic groove, short ribs, and hypoplastic iliac wings compared to nonsense variants. They were also significantly associated with delayed fontanelle closure, metopic synostosis, hypertelorism, limited shoulder abduction, pubic symphysis abnormalities, and hypoplastic iliac wings compared to in-frame variants found in other regions. These findings underscore the critical role of the RHD, with missense RHD variants having a more severe impact than nonsense and other in-frame variants. Additionally, in-frame insertions and deletions in RUNX2 were associated with fewer CCD features, compared to missense, frameshift, and nonsense variants. Null variants in the NLS region exhibited weaker associations with delayed fontanelle closure, supernumerary teeth, Wormian bones, and femoral head hypoplasia than variants in other regions. Moreover, the NLS variants did not consistently alter nuclear localization, questioning the role of NLS region in nuclear import. In summary, this comprehensive review significantly advances our understanding of CCD, facilitating improved phenotype-genotype correlations, enhanced clinical management, and a deeper insight into RUNX2 functional domains. This knowledge has the potential to guide the development of novel therapeutic targets for skeletal disorders.
锁骨颅骨发育不全(CCD)是一种罕见的遗传性疾病,其特征为骨骼异常和牙齿畸形,主要由RUNX2基因变异引起。了解RUNX2变异的范围及其对CCD表型的影响对于准确诊断和制定管理策略至关重要。本系统综述旨在全面分析CCD患者中RUNX2变异的基因型和表型谱,评估其在功能区域的分布,并研究基因型与表型的相关性。该综述纳入了103篇文章中报道的569个变异和453例CCD患者。在569个变异中,框内变异占48.68%,而无效变异占51.32%。在位置方面,RUNX2变异主要位于RHD区域(55.54%),其次是PST区域(16.34%)、NMTS区域(6.33%)、QA区域(4.75%)、VWRPY区域(1.23%)和NLS区域(1.41%),而10.19%位于非编码区域。框内变异主要发生在RHD区域(90.97%),而无效变异在RUNX2的各个区域均有发现。数据分析揭示了CCD患者变异位置与特定骨骼特征之间的相关性。与无义变异相比,主要位于功能关键的RHD区域内的错义变异与多生牙、巨头畸形、额缝、短肋骨和髂骨翼发育不全显著相关。与在其他区域发现的框内变异相比,它们还与囟门闭合延迟、额缝早闭、眼距过宽、肩关节外展受限、耻骨联合异常和髂骨翼发育不全显著相关。这些发现强调了RHD的关键作用,错义RHD变异的影响比无义变异和其他框内变异更为严重。此外,与错义、移码和无义变异相比,RUNX2中的框内插入和缺失与较少的CCD特征相关。NLS区域的无效变异与囟门闭合延迟、多生牙、缝间骨和股骨头发育不全的关联比其他区域的变异弱。此外,NLS变异并未一致地改变核定位,这对NLS区域在核输入中的作用提出了质疑。总之,这一全面综述显著推进了我们对CCD的理解,有助于改善表型-基因型相关性、加强临床管理,并更深入地了解RUNX2功能域。这些知识有可能指导骨骼疾病新型治疗靶点的开发。