La Rosa Alessandro, Covone Angela Elvira, Coviello Domenico, Arrigo Serena, Ferro Jacopo, Gandullia Paolo, Madeo Annalisa
Paediatric Gastroenterology and Digestive Endoscopy Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy.
Laboratory of Human Genetics, IRCCS Istituto Giannina Gaslini, Genoa, Italy.
Case Reports Hepatol. 2024 Nov 25;2024:3815089. doi: 10.1155/crhe/3815089. eCollection 2024.
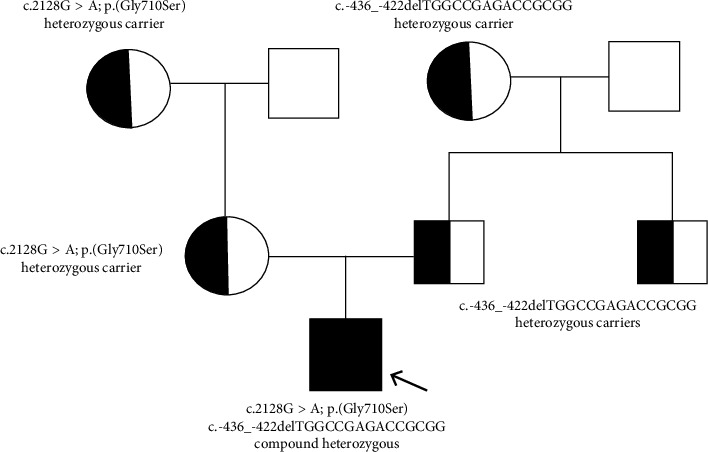
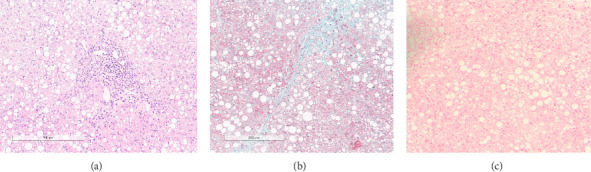
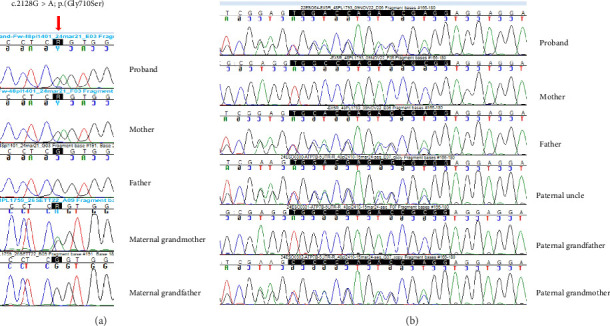
Wilson's disease (WD) is a rare autosomal recessive disorder caused by mutations in the ATP7B gene, resulting in copper accumulation. Symptoms rarely appear before the age of 5, almost never before 3. The phenotypic variability of WD suggests the presence of modifying factors, making early diagnosis challenging. We present a case of symptomatic WD in a toddler, emphasizing the importance of considering WD in differential diagnoses and exploring genetic modifiers influencing disease onset. Clinical and laboratory assessments, including liver biopsy, were performed on a 4.2-year-old boy presenting with hypertransaminasemia and mild hepatomegaly. Histological evaluation revealed chronic hepatitis with fibrosis and severe steatosis, indicating long-standing active disease. Genetic analysis identified a missense variant and a 15-nucleotide deletion in the 5' UTR promoter region of the ATP7B gene, confirming the WD diagnosis. Additionally, homozygosity for the HFE H63D variant was detected, with transferrin saturations at the upper limit of normal. The patient's clinical management included a trial of D-penicillamine, discontinued due to side effects, followed by successful zinc acetate therapy. This case underscores the consideration of WD in the differential diagnosis of toddlers. The Ferenci-Leipzig score remains a valid diagnostic tool for WD even in the presence of a single ATP7B variant, although extended genetic analysis should still be considered. Normal ceruloplasmin levels do not rule out WD. Environmental, epigenetic, and genetic factors appear to influence the WD phenotype; HFE variants may act as modifiers given the link between iron and copper homeostasis, possibly explaining the early symptomatic onset in our patient.
威尔逊病(WD)是一种罕见的常染色体隐性疾病,由ATP7B基因突变引起,导致铜蓄积。症状很少在5岁前出现,3岁前几乎从不出现。WD的表型变异性提示存在修饰因子,这使得早期诊断具有挑战性。我们报告一例幼儿期出现症状的WD病例,强调在鉴别诊断中考虑WD以及探索影响疾病发病的遗传修饰因子的重要性。对一名4.2岁出现高转氨酶血症和轻度肝肿大的男孩进行了临床和实验室评估,包括肝活检。组织学评估显示为伴有纤维化和严重脂肪变性的慢性肝炎,提示存在长期活动性疾病。基因分析在ATP7B基因的5'UTR启动子区域鉴定出一个错义变异和一个15核苷酸缺失,证实了WD诊断。此外,检测到HFE H63D变异的纯合性,转铁蛋白饱和度处于正常上限。患者的临床治疗包括试用D-青霉胺,但因副作用停药,随后成功接受醋酸锌治疗。该病例强调在幼儿鉴别诊断中考虑WD。即使存在单个ATP7B变异,Ferenci-Leipzig评分仍是WD的有效诊断工具,不过仍应考虑进行扩展基因分析。正常的铜蓝蛋白水平不能排除WD。环境、表观遗传和遗传因素似乎会影响WD表型;鉴于铁与铜稳态之间的联系,HFE变异可能作为修饰因子,这可能解释了我们患者出现早期症状性发病的原因。