Bull Courtney, Byrne Ryan M, Fisher Natalie C, Corry Shania M, Amirkhah Raheleh, Edwards Jessica, Hillson Lily V S, Lawler Mark, Ryan Aideen E, Lamrock Felicity, Dunne Philip D, Malla Sudhir B
The Patrick G Johnston Centre for Cancer Research, Queen's University Belfast, Belfast, UK.
School of Cancer Sciences, University of Glasgow, Glasgow, UK.
Sci Rep. 2024 Dec 4;14(1):30202. doi: 10.1038/s41598-024-80534-8.
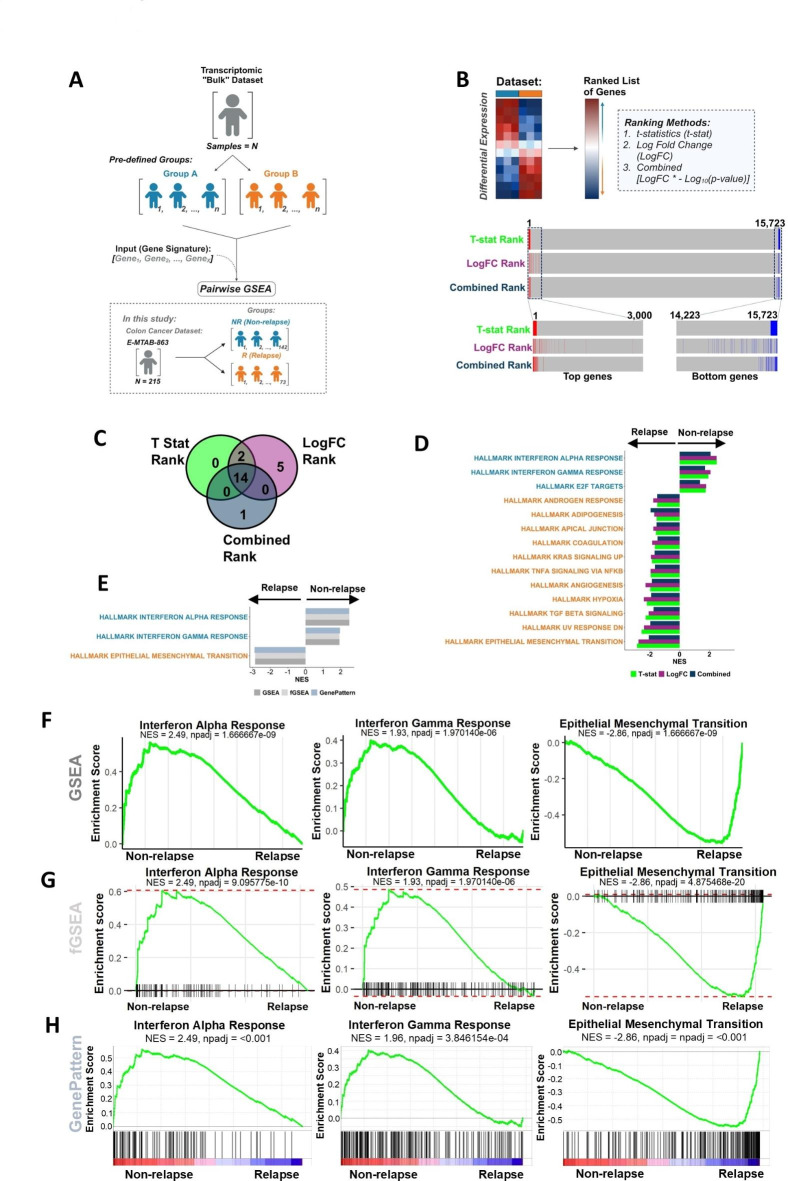
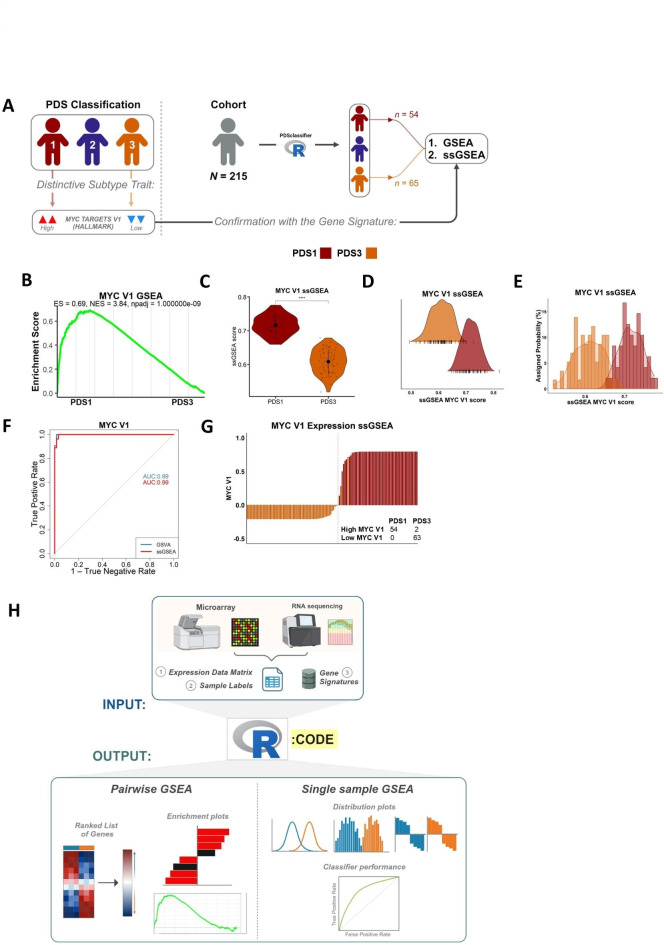
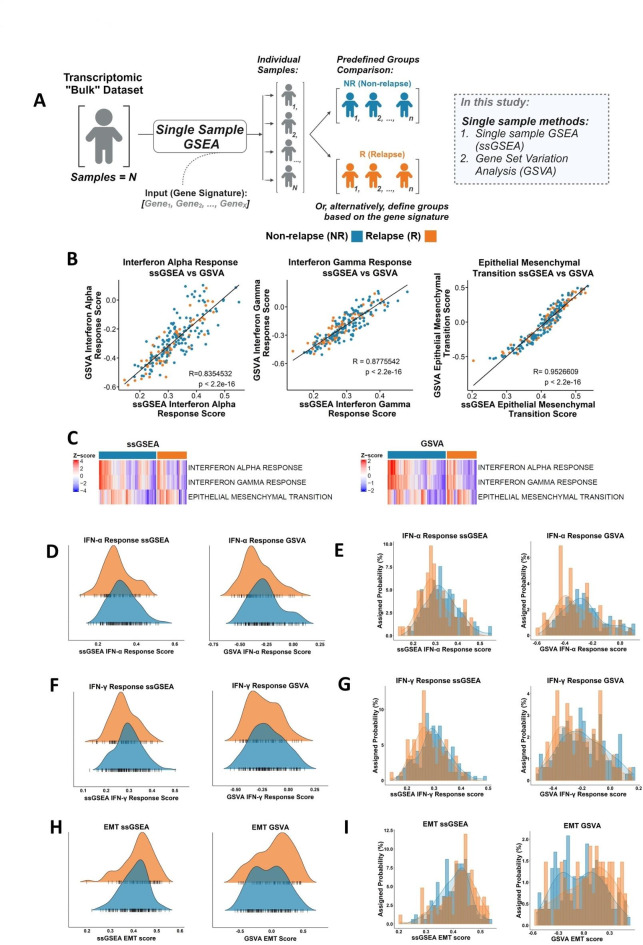
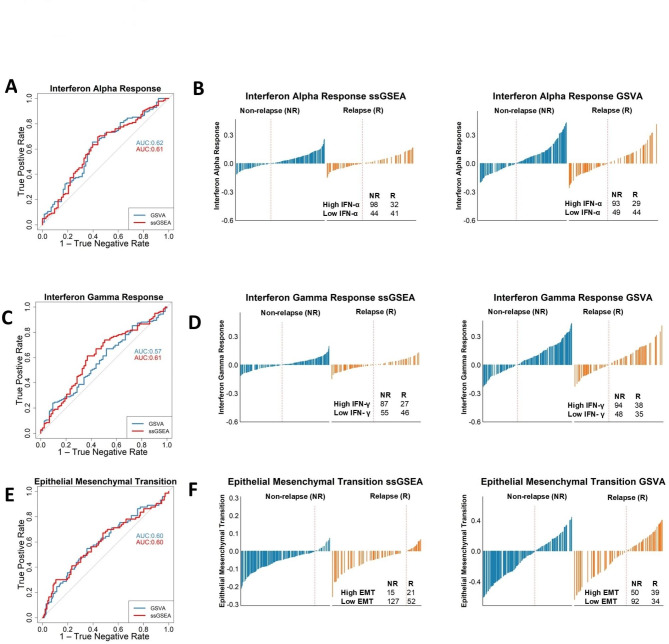
Gene set enrichment analysis (GSEA) tools can identify biological insights within gene expression-based studies. Although their statistical performance has been compared, the downstream biological implications that arise when choosing between the range of pairwise or single sample forms of GSEA methods remain understudied. We compare the statistical and biological results obtained from various pre-ranking methods/options for pairwise GSEA, followed by a stand-alone comparison of GSEA, single sample GSEA (ssGSEA) and gene set variation analysis (GSVA). Pairwise GSEA and fGSEA provide similar results when deployed using a range of gene pre-ranking methods. However, pairwise GSEA can overgeneralise biological enrichment, as when the most statistically significant signatures were assessed using single sample approaches, there was a complete absence of biological distinction between these groups. To avoid these issues, we developed a new dualGSEA tool, which provides users with multiple statistics and visuals to aid interpretation of results. This new tool removes the possibility of users inadvertently interpreting statistical findings as equating to biological distinction between samples within groups-of-interest. dualGSEA provides a more robust basis for discovery research, one which allows user to compare both statistical significance alongside biological distinctions in their data.
基因集富集分析(GSEA)工具能够在基于基因表达的研究中识别生物学见解。尽管已对其统计性能进行了比较,但在GSEA方法的成对或单样本形式范围内进行选择时所产生的下游生物学影响仍未得到充分研究。我们比较了从成对GSEA的各种预排序方法/选项中获得的统计和生物学结果,随后对GSEA、单样本GSEA(ssGSEA)和基因集变异分析(GSVA)进行了单独比较。当成对GSEA使用一系列基因预排序方法进行部署时,成对GSEA和fGSEA提供了相似的结果。然而,成对GSEA可能会过度概括生物学富集情况,因为当使用单样本方法评估最具统计学意义的特征时,这些组之间完全没有生物学差异。为避免这些问题,我们开发了一种新的dualGSEA工具,它为用户提供多种统计信息和可视化手段,以帮助解释结果。这个新工具消除了用户无意中将统计结果解释为等同于感兴趣组内样本之间生物学差异的可能性。dualGSEA为发现性研究提供了一个更可靠的基础,它允许用户在数据中同时比较统计显著性和生物学差异。