Ormond Cathal, Ryan Niamh M, Cap Mathieu, Byerley William, Corvin Aiden, Heron Elizabeth A
Neuropsychiatric Genetics Research Group, Department of Psychiatry, Trinity Centre for Health Sciences, Trinity College Dublin, St James's Hospital, Dublin 8, Ireland.
Department of Psychiatry and Behavioral Sciences, University of California, 1550 Fourth Street, San Francisco, CA 94158, United States.
Brief Bioinform. 2024 Nov 22;26(1). doi: 10.1093/bib/bbae624.
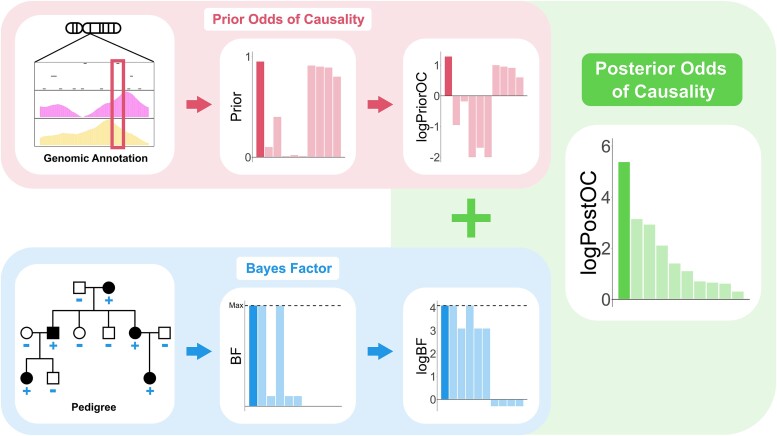
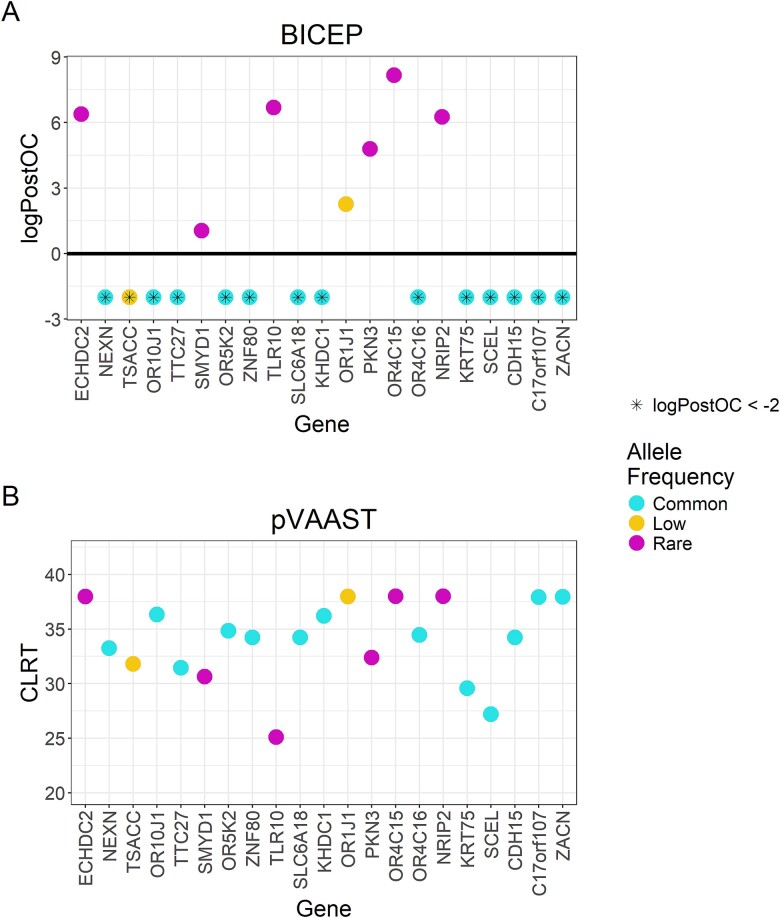
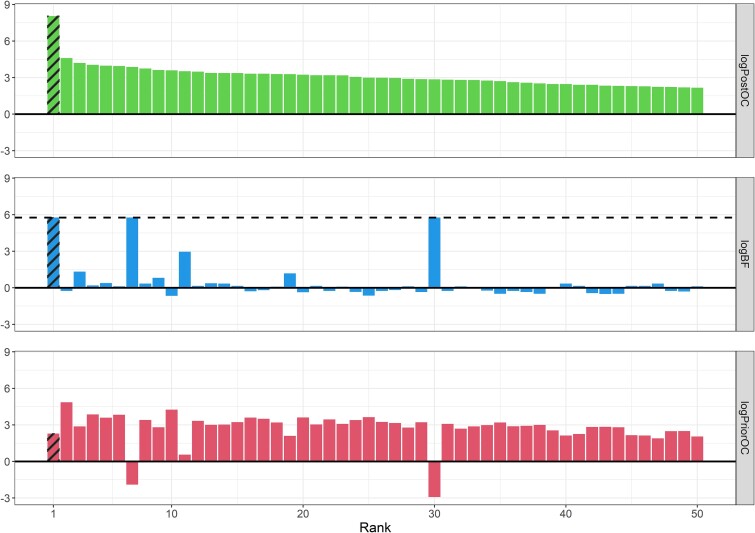
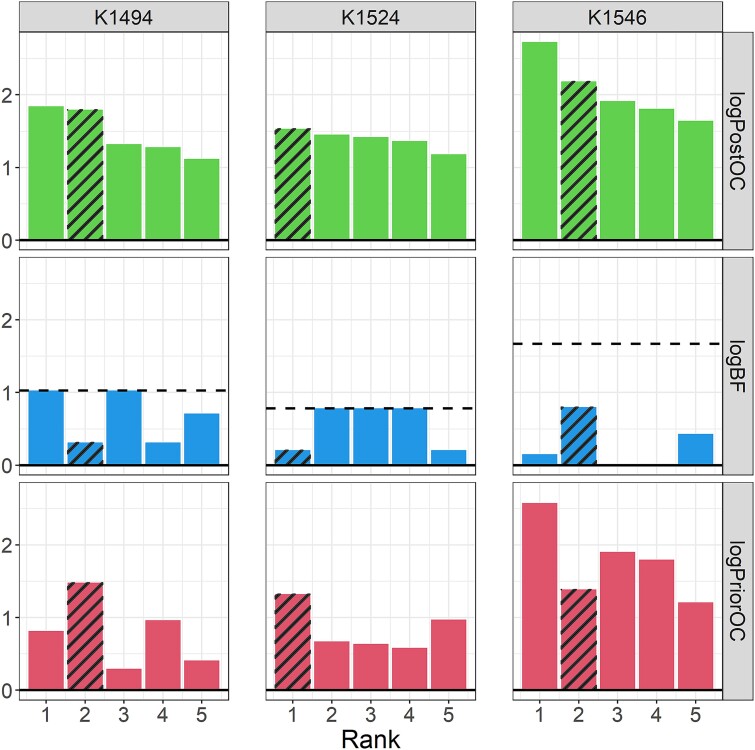
Next-generation sequencing is widely applied to the investigation of pedigree data for gene discovery. However, identifying plausible disease-causing variants within a robust statistical framework is challenging. Here, we introduce BICEP: a Bayesian inference tool for rare variant causality evaluation in pedigree-based cohorts. BICEP calculates the posterior odds that a genomic variant is causal for a phenotype based on the variant cosegregation as well as a priori evidence such as deleteriousness and functional consequence. BICEP can correctly identify causal variants for phenotypes with both Mendelian and complex genetic architectures, outperforming existing methodologies. Additionally, BICEP can correctly down-weight common variants that are unlikely to be involved in phenotypic liability in the context of a pedigree, even if they have reasonable cosegregation patterns. The output metrics from BICEP allow for the quantitative comparison of variant causality within and across pedigrees, which is not possible with existing approaches.
下一代测序被广泛应用于家系数据研究以发现基因。然而,在强大的统计框架内识别合理的致病变异具有挑战性。在此,我们引入了BICEP:一种用于基于家系队列的罕见变异因果关系评估的贝叶斯推理工具。BICEP基于变异共分离以及有害性和功能后果等先验证据,计算基因组变异导致某一表型的后验优势比。BICEP能够正确识别具有孟德尔遗传和复杂遗传结构的表型的因果变异,性能优于现有方法。此外,即使常见变异在家系背景下具有合理的共分离模式,但如果它们不太可能与表型易感性相关,BICEP也能够正确地降低其权重。BICEP的输出指标允许对家系内部和家系之间的变异因果关系进行定量比较,而现有方法无法做到这一点。