Twa Guy M, Phillips Robert A, Robinson Nathaniel J, Day Jeremy J
Department of Neurobiology, University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Present affiliation: Lieber Institute for Brain Development, Johns Hopkins Medical Campus, Baltimore, MD 21205, USA.
bioRxiv. 2024 Dec 3:2024.11.29.626066. doi: 10.1101/2024.11.29.626066.
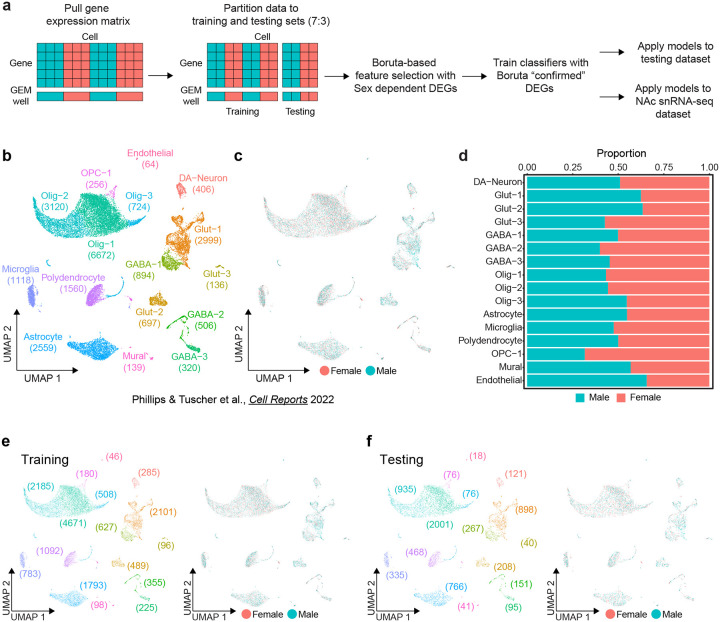
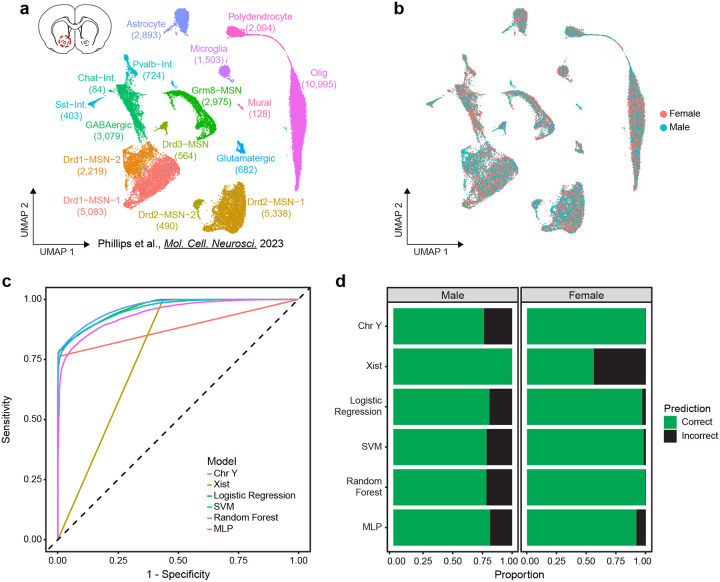
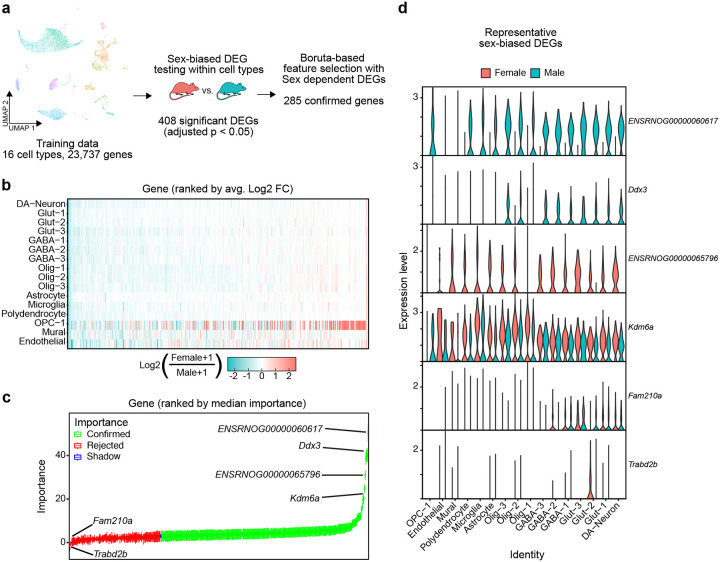
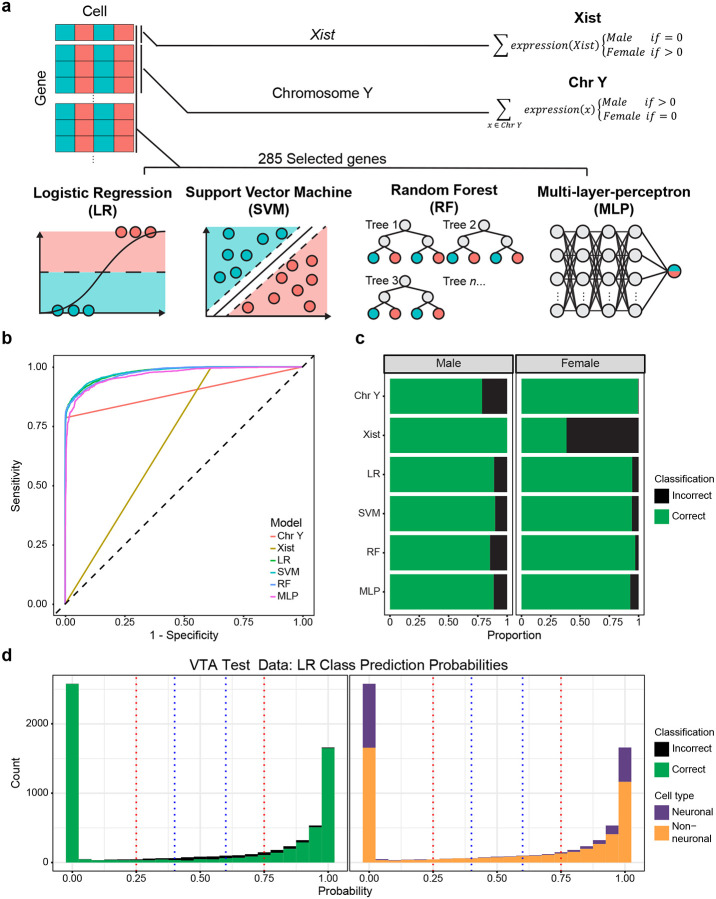
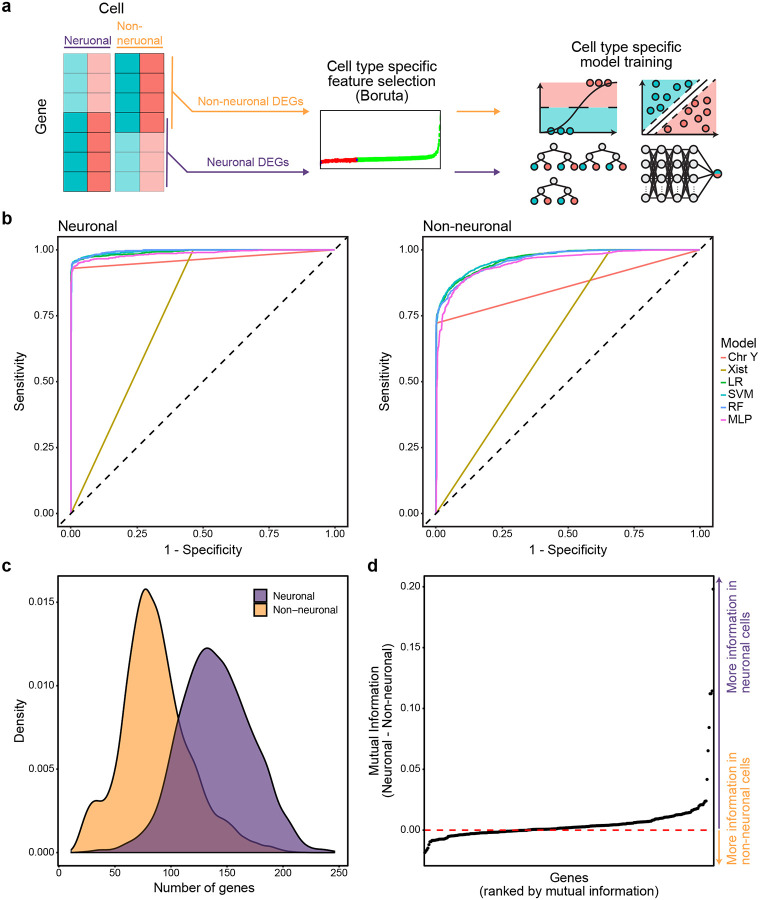
Single nucleus RNA sequencing (snRNA-seq) technology offers unprecedented resolution for studying cell type-specific gene expression patterns. However, snRNA-seq poses high costs and technical limitations, often requiring the pooling of independent biological samples and loss of individual sample-level data. Deconvolution of sample identity using inherent features would enable the incorporation of pooled barcoding and sequencing protocols, thereby increasing data throughput and analytical sample size without requiring increases in experimental sample size and sequencing costs. In this study, we demonstrate a proof of concept that sex-dependent gene expression patterns can be leveraged for the deconvolution of pooled snRNA-seq data. Using previously published snRNA-seq data from the rat ventral tegmental area, we trained a range of machine learning models to classify cell sex using genes differentially expressed in cells from male and female rats. Models that used sex-dependent gene expression predicted cell sex with high accuracy (90-92%) and outperformed simple classification models using only sex chromosome gene expression (69-89%). The generalizability of these models to other brain regions was assessed using an additional published data set from the rat nucleus accumbens. Within this data set, model performance remained highly accurate in cell sex classification (89-90% accuracy) with no additional re-training. This work provides a model for future snRNA-seq studies to perform sample deconvolution using a two-sex pooled sample sequencing design and benchmarks the performance of various machine learning approaches to deconvolve sample identification from inherent sample features.
单细胞核RNA测序(snRNA-seq)技术为研究细胞类型特异性基因表达模式提供了前所未有的分辨率。然而,snRNA-seq成本高昂且存在技术限制,通常需要将独立的生物样本合并,从而导致个体样本水平的数据丢失。利用固有特征对样本身份进行反卷积,将能够采用合并条形码和测序方案,从而在不增加实验样本量和测序成本的情况下提高数据通量和分析样本量。在本研究中,我们证明了一个概念验证,即性别依赖性基因表达模式可用于对合并的snRNA-seq数据进行反卷积。利用先前发表的大鼠腹侧被盖区的snRNA-seq数据,我们训练了一系列机器学习模型,使用雄性和雌性大鼠细胞中差异表达的基因来分类细胞性别。使用性别依赖性基因表达的模型能够高精度地预测细胞性别(90-92%),并且优于仅使用性染色体基因表达的简单分类模型(69-89%)。使用来自大鼠伏隔核的另一个已发表数据集评估了这些模型对其他脑区的通用性。在该数据集中,模型在细胞性别分类中的性能保持高度准确(准确率89-90%),无需额外重新训练。这项工作为未来的snRNA-seq研究提供了一个模型,以使用两性合并样本测序设计进行样本反卷积,并对各种机器学习方法从固有样本特征中反卷积样本识别的性能进行了基准测试。