Oba Fumiyasu, Nagai Takayuki, Katsube Ryoji, Mochizuki Yasuhide, Tsuji Masatake, Deffrennes Guillaume, Hanzawa Kota, Nakano Akitoshi, Takahashi Akira, Terayama Kei, Tamura Ryo, Hiramatsu Hidenori, Nose Yoshitaro, Taniguchi Hiroki
Laboratory for Materials and Structures, Institute of Innovative Research, Tokyo Institute of Technology, Yokohama, Japan.
Department of Physics, Nagoya University, Nagoya, Japan.
Sci Technol Adv Mater. 2024 Nov 4;25(1):2423600. doi: 10.1080/14686996.2024.2423600. eCollection 2024.
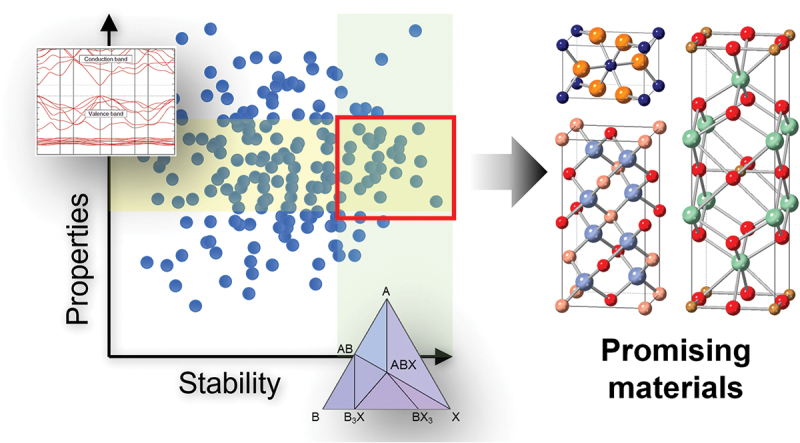
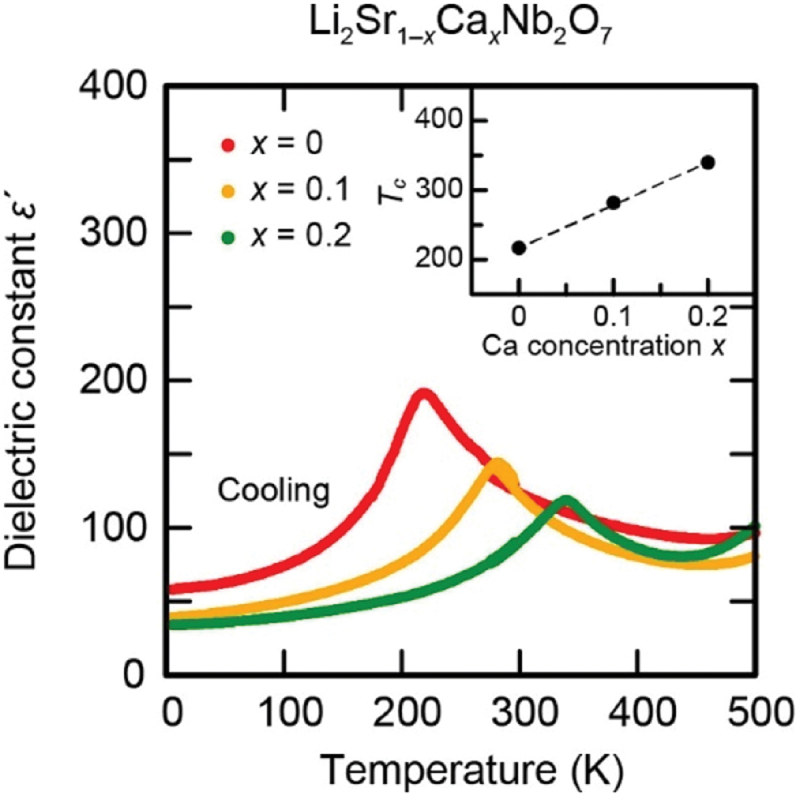
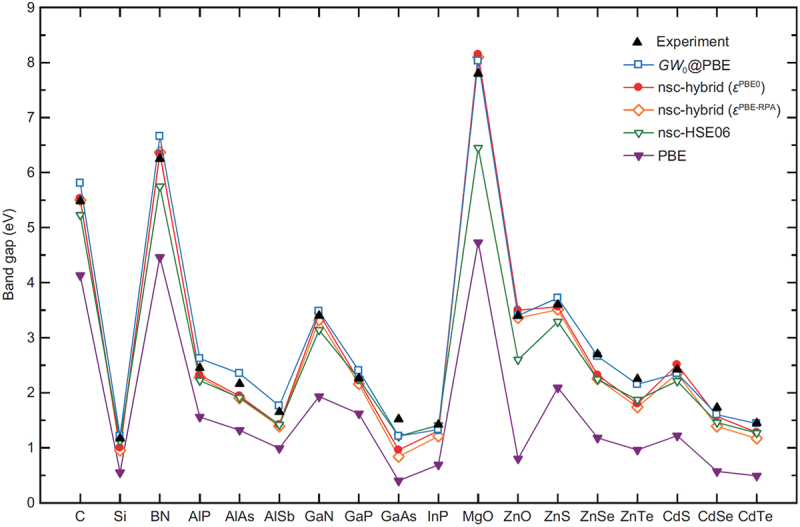
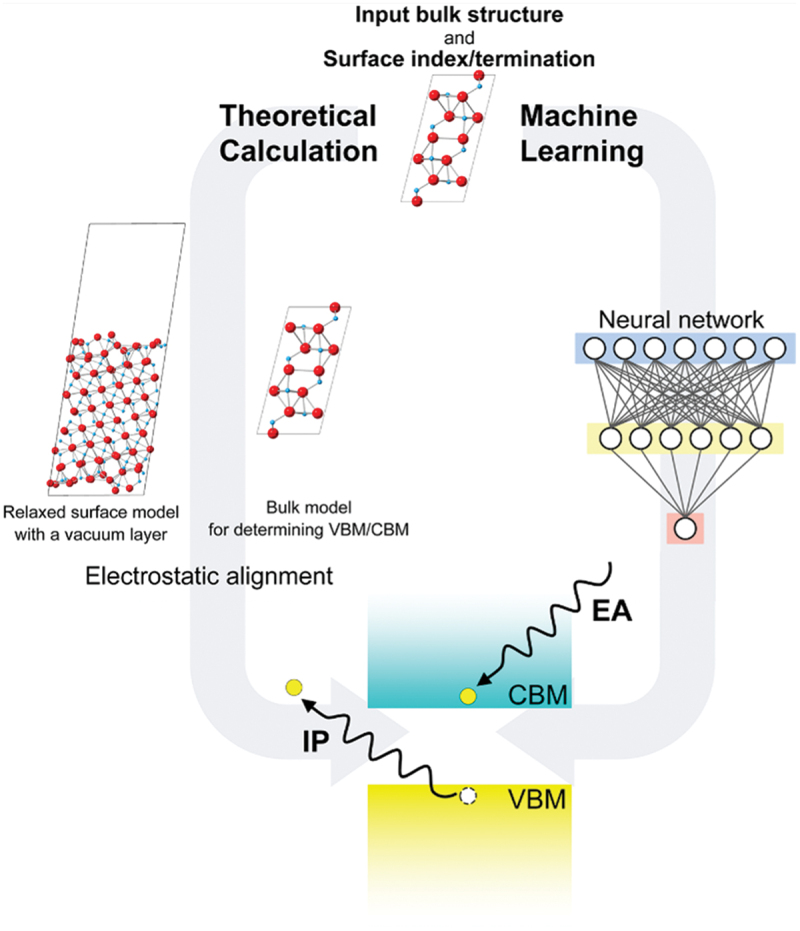
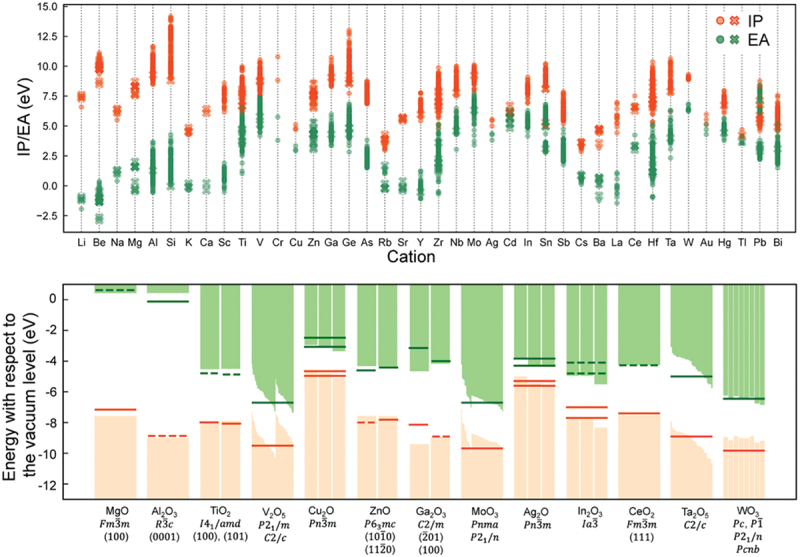
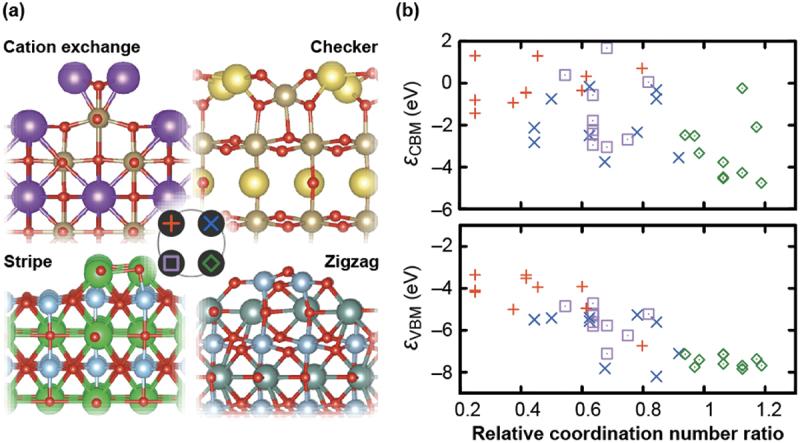
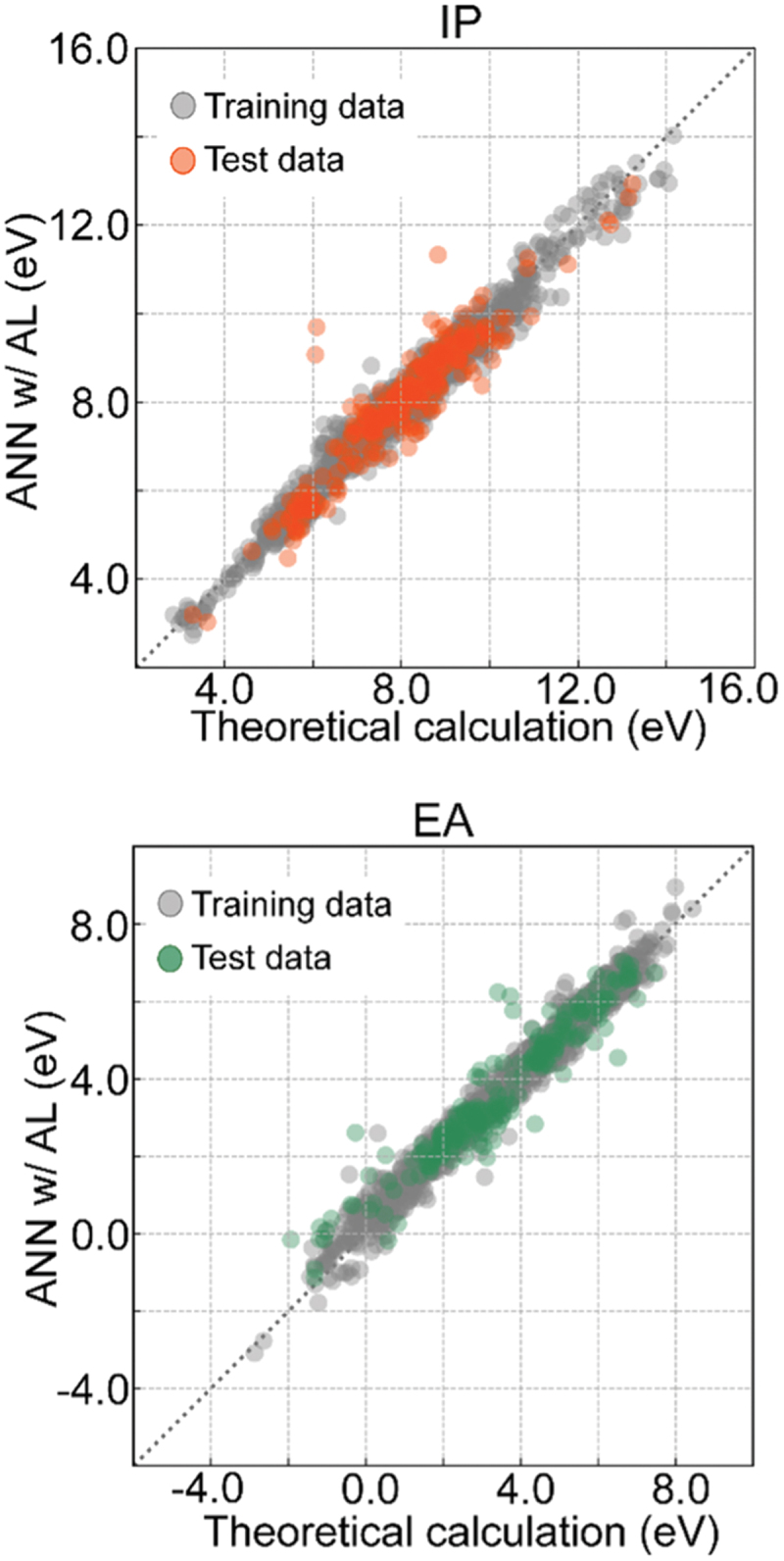
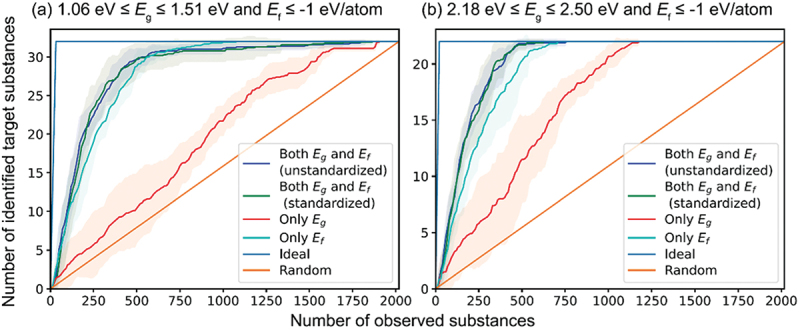
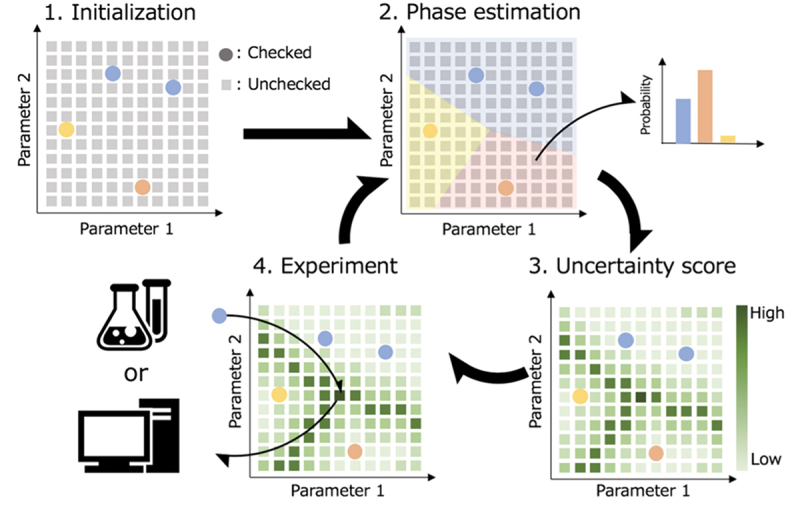
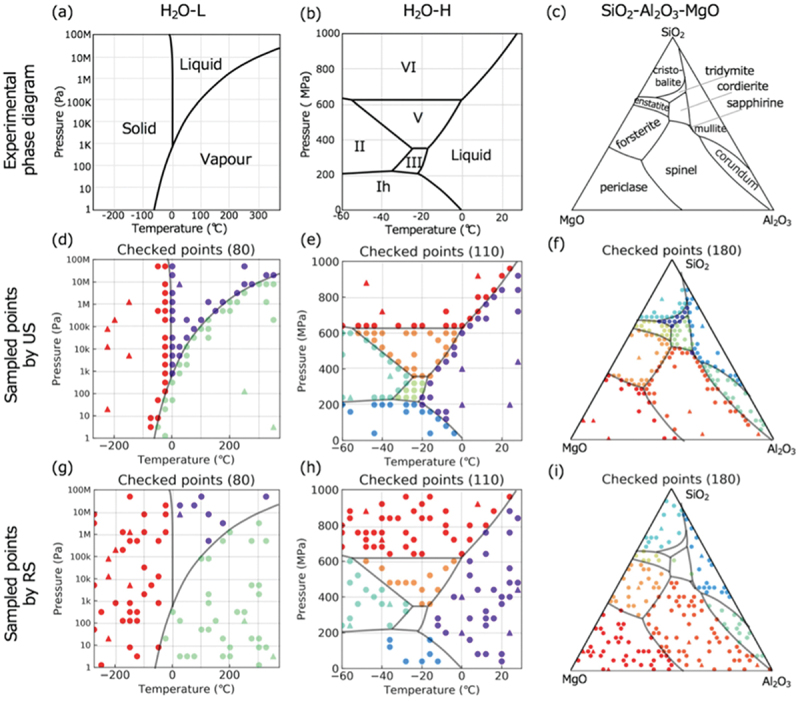
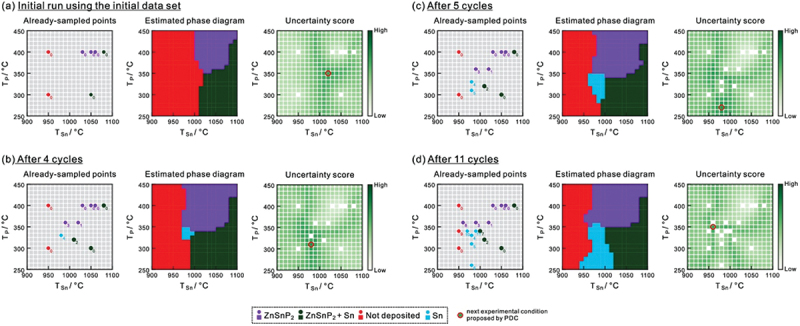
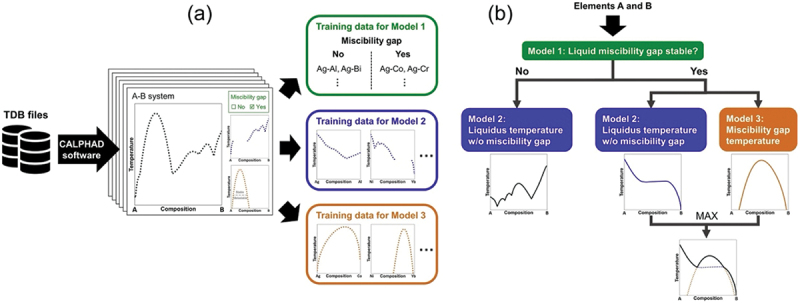
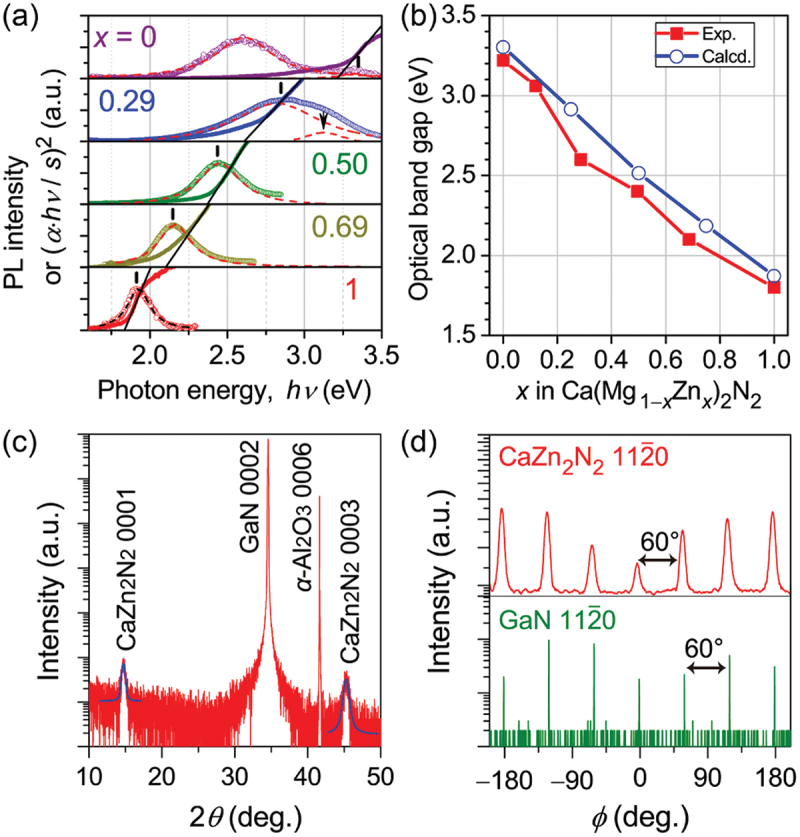
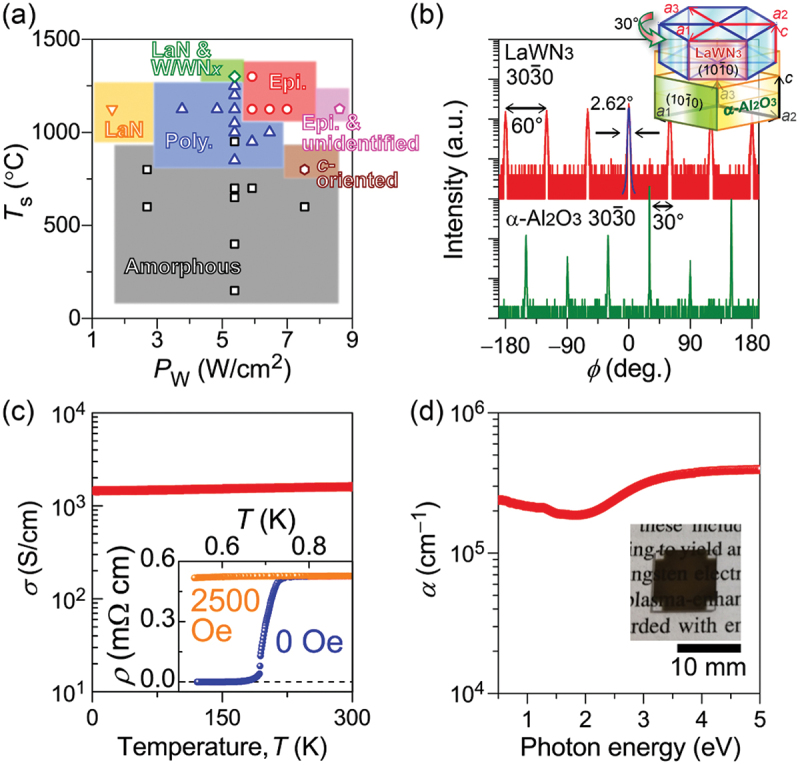
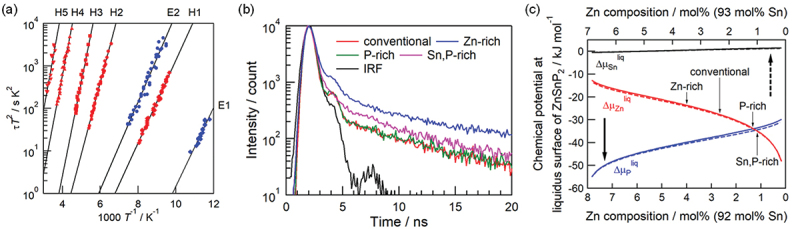
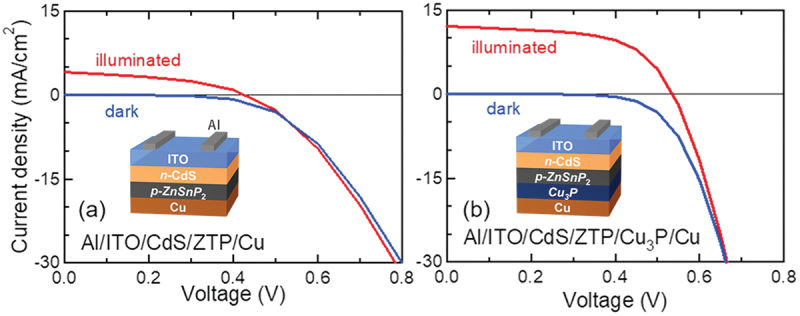
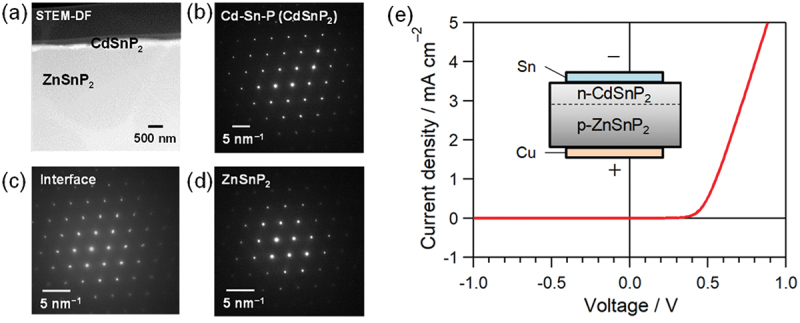
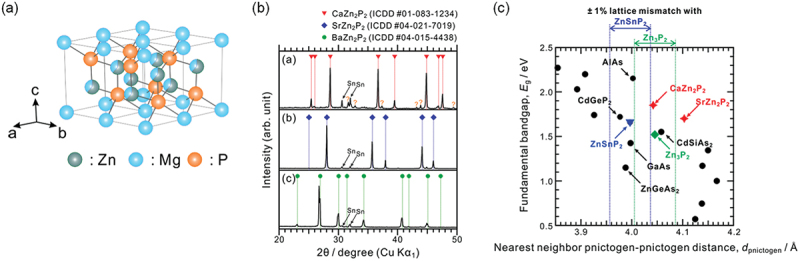
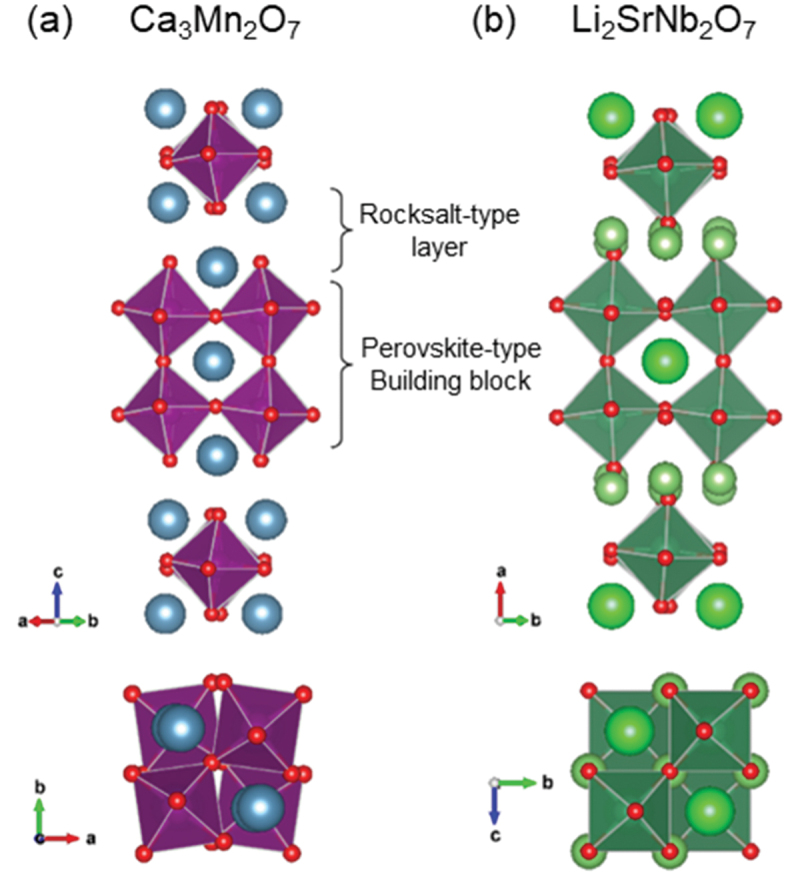
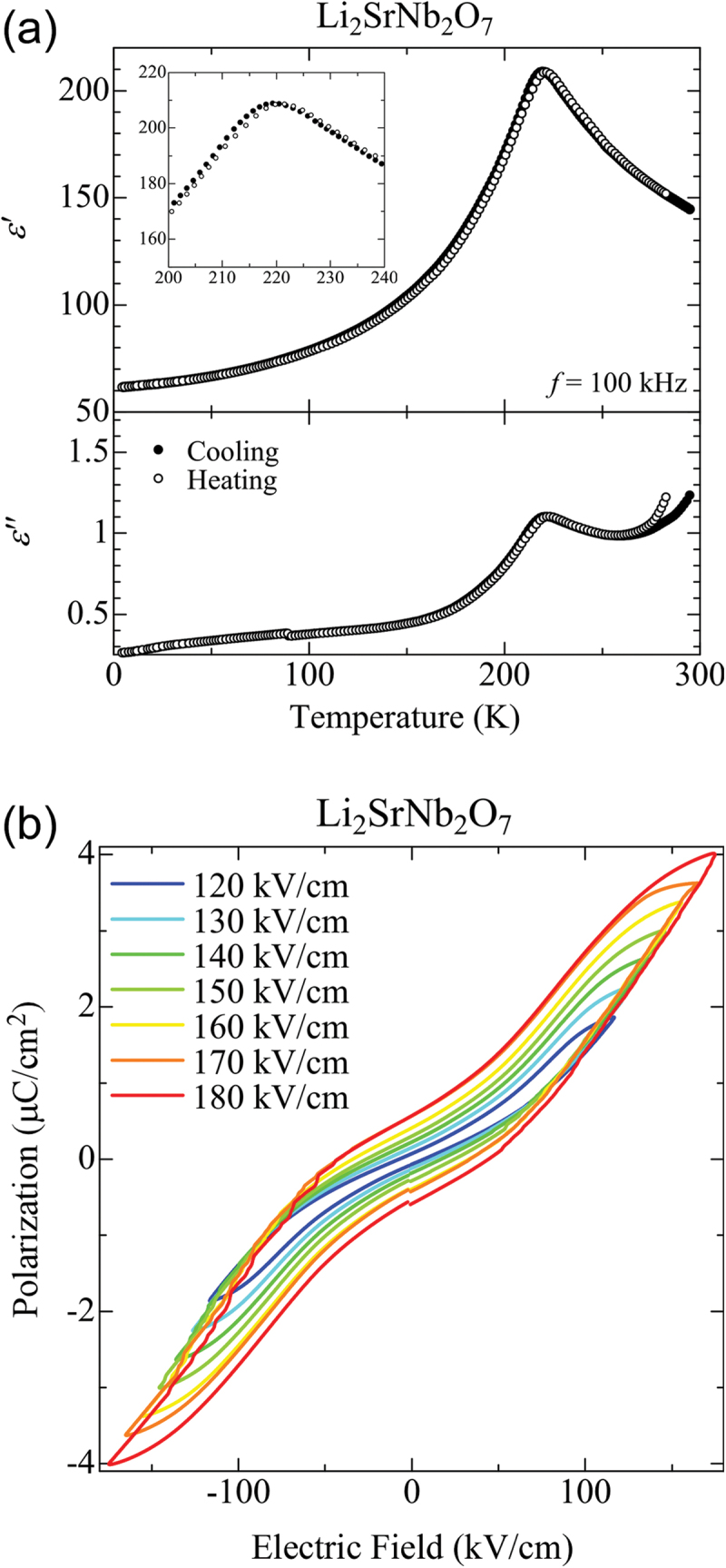
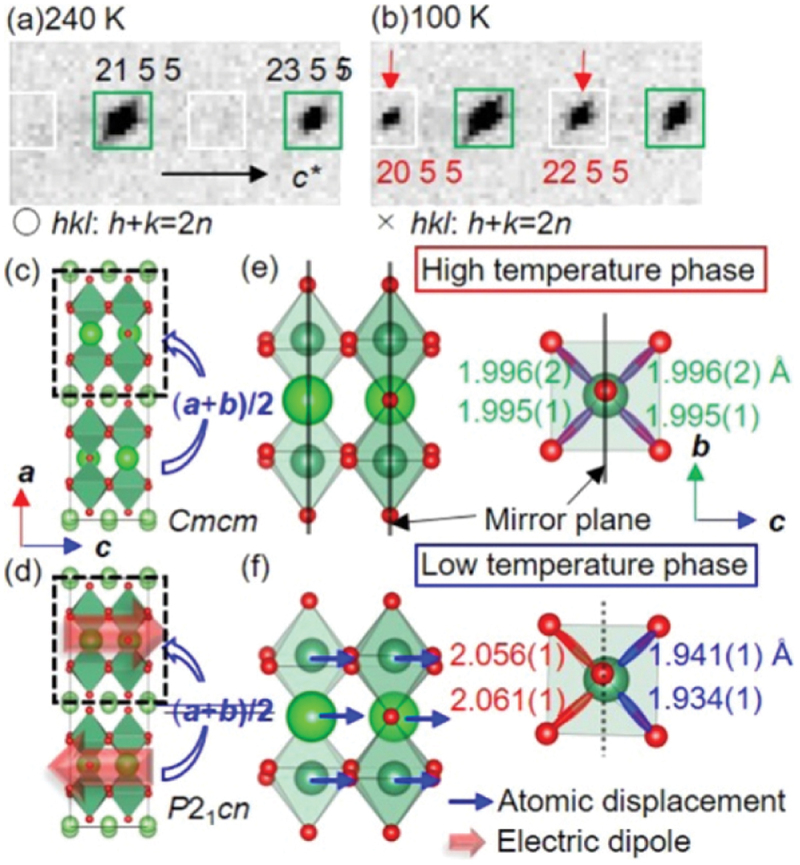
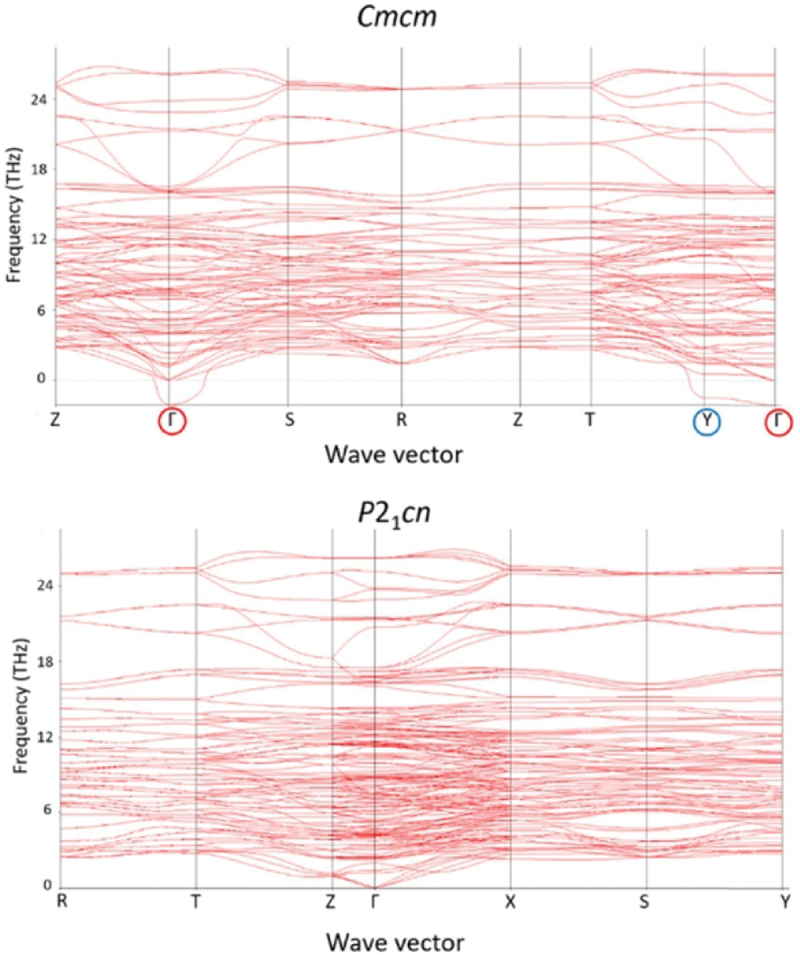
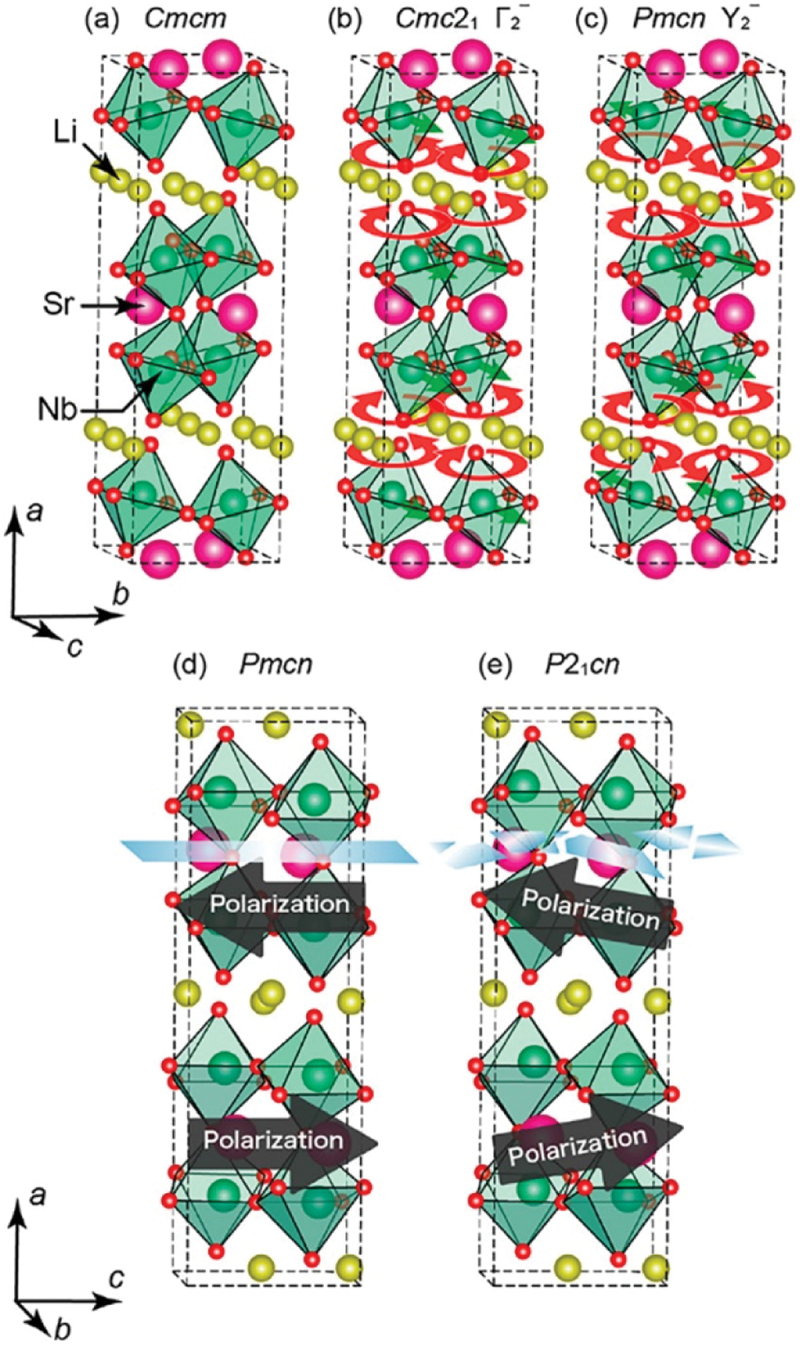
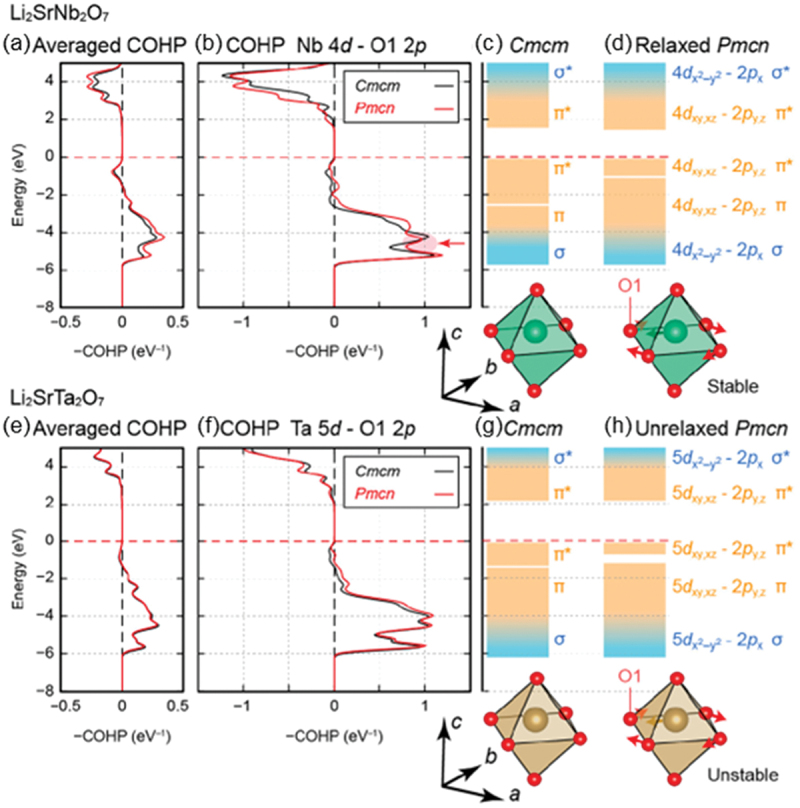
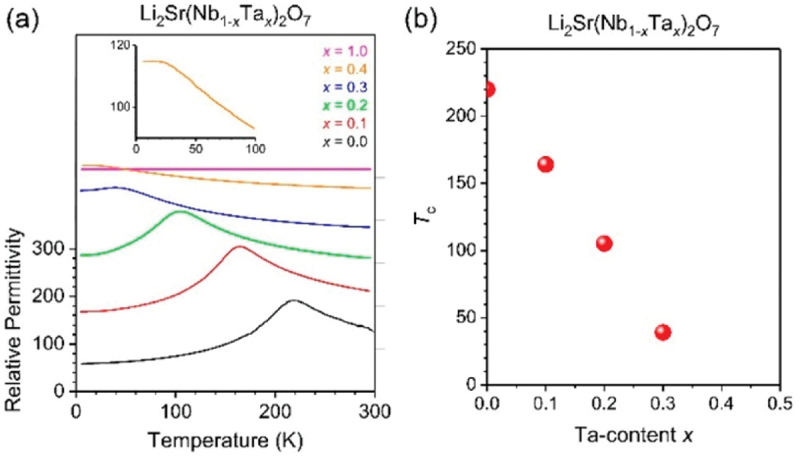
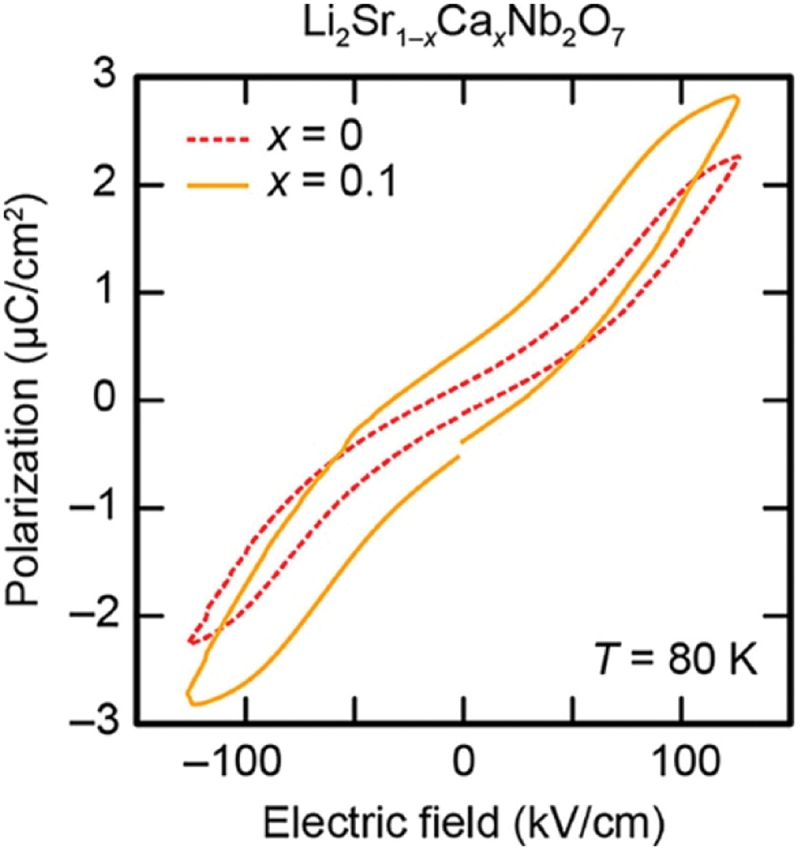
Computational approaches using theoretical calculations and data scientific methods have become increasingly important in materials science and technology, with the development of relevant methodologies and algorithms, the availability of large materials data, and the enhancement of computer performance. As reviewed herein, we have developed computational methods for the design and prediction of inorganic materials with a particular focus on the exploration of semiconductors and dielectrics. High-throughput first-principles calculations are used to systematically and accurately predict the local atomic and electronic structures of polarons, point defects, surfaces, and interfaces, as well as bulk fundamental properties. Machine learning techniques are utilized to efficiently predict various material properties, construct phase diagrams, and search for materials satisfying target properties. These computational approaches have elucidated the mechanisms behind material functionalities and explored promising materials in combination with synthesis, characterization, and device fabrication. Examples include the development of ternary nitride semiconductors for potential optoelectronic and photovoltaic applications, the exploration of phosphide semiconductors and the optimization of heterointerfaces toward the improvement of phosphide-based photovoltaic cells, and the discovery of ferroelectricity in layered perovskite oxides and the theoretical understanding of its origin, all of which demonstrate the effectiveness of our computer-aided materials research.
随着相关方法和算法的发展、大量材料数据的可得性以及计算机性能的提升,使用理论计算和数据科学方法的计算方法在材料科学与技术中变得越来越重要。如本文所述,我们已经开发了用于无机材料设计和预测的计算方法,特别关注半导体和电介质的探索。高通量第一性原理计算用于系统且准确地预测极化子、点缺陷、表面和界面的局部原子和电子结构,以及体相基本性质。利用机器学习技术高效预测各种材料性质、构建相图并搜索满足目标性质的材料。这些计算方法阐明了材料功能背后的机制,并结合合成、表征和器件制造探索了有前景的材料。实例包括开发用于潜在光电子和光伏应用的三元氮化物半导体、探索磷化物半导体以及优化异质界面以改进基于磷化物的光伏电池,以及在层状钙钛矿氧化物中发现铁电性并对其起源进行理论理解,所有这些都证明了我们计算机辅助材料研究的有效性。