Sarashetti Prasad, Lipovac Josipa, Tomas Filip, Šikić Mile, Liu Jianjun
Laboratory of Human Genomics, Genome Institute of Singapore, A*STAR, Singapore, Singapore.
Laboratory for Bioinformatics and Computational Biology, Faculty of Electrical Engineering and Computing, University of Zagreb, Zagreb, Croatia.
Genome Biol. 2024 Dec 18;25(1):312. doi: 10.1186/s13059-024-03452-y.
Long-read technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) have transformed genomics research by providing diverse data types like HiFi, Duplex, and ultra-long ONT. Despite recent strides in achieving haplotype-phased gapless genome assemblies using long-read technologies, concerns persist regarding the representation of genetic diversity, prompting the development of pangenome references. However, pangenome studies face challenges related to data types, volumes, and cost considerations for each assembled genome, while striving to maintain sensitivity. The absence of comprehensive guidance on optimal data selection exacerbates these challenges.
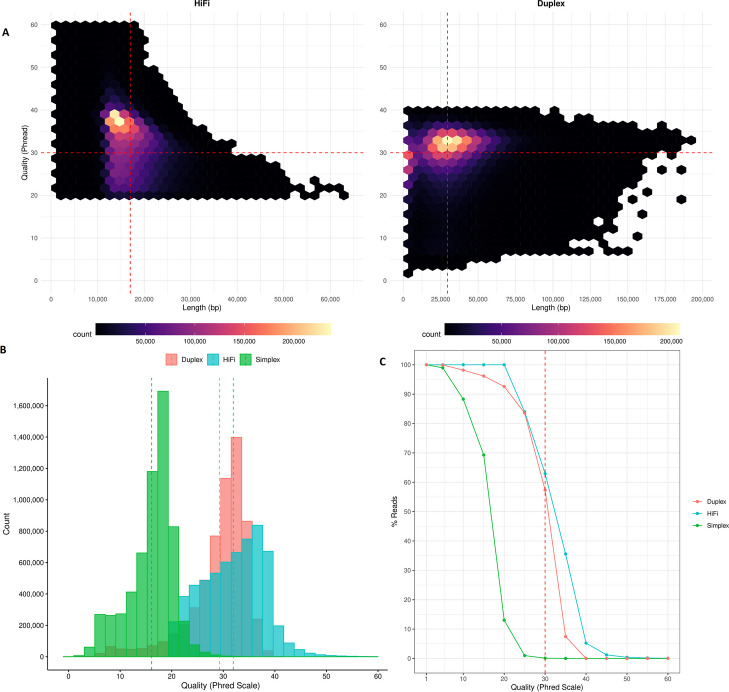
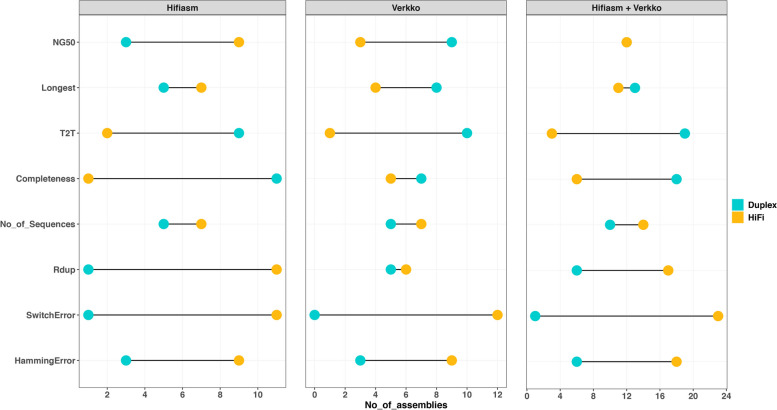
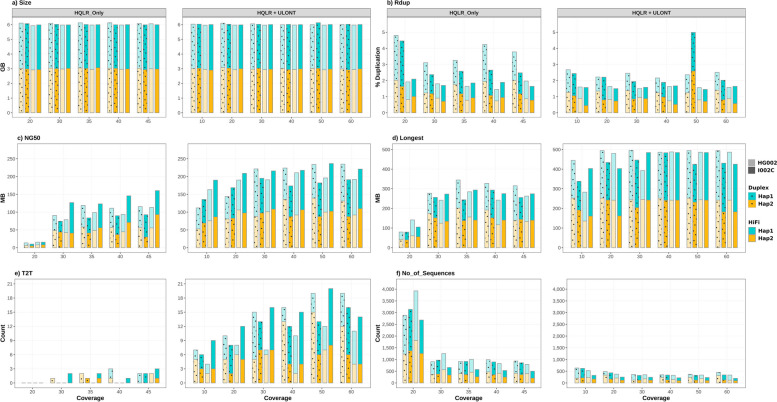
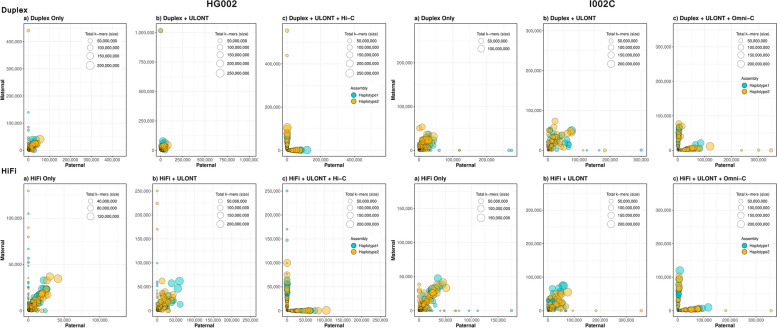
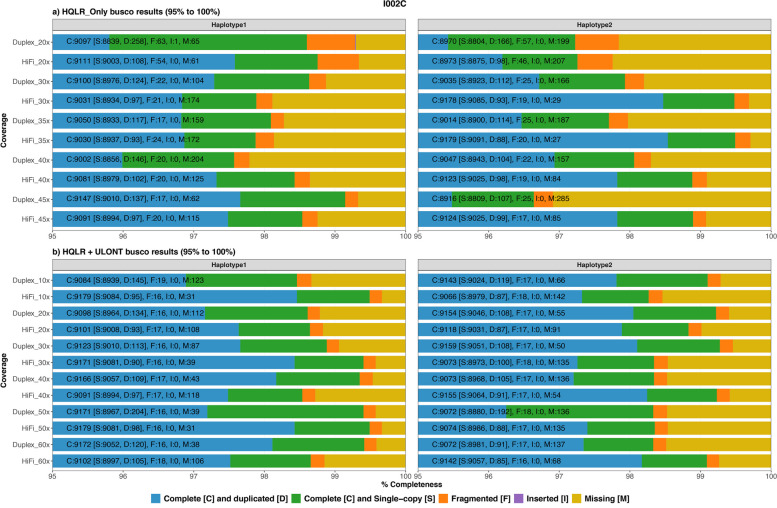
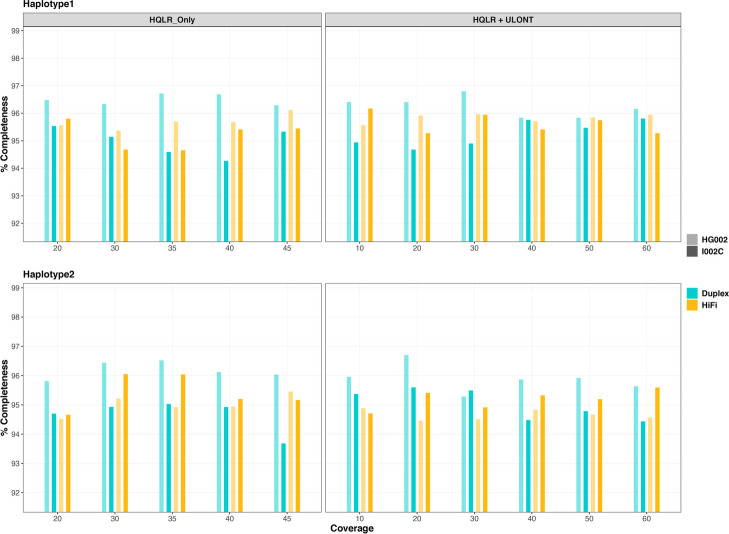
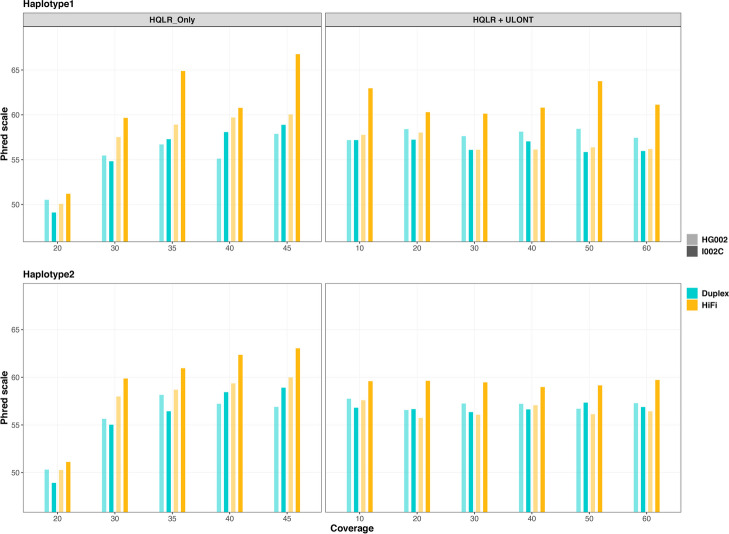
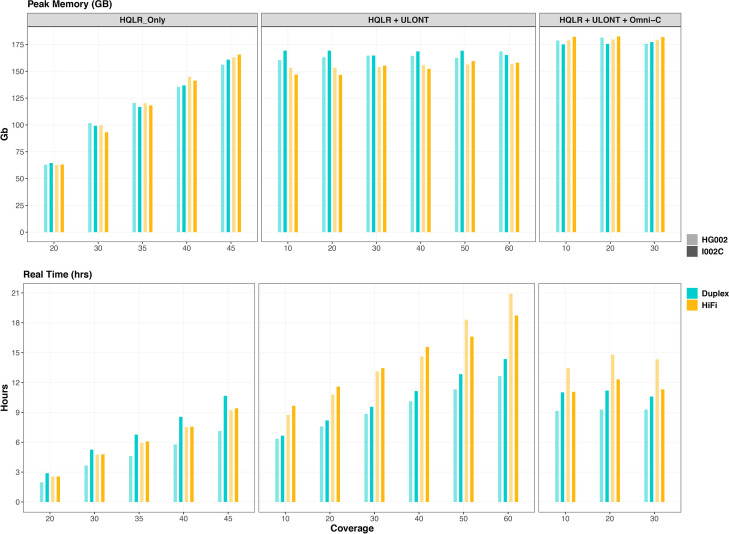
Our study evaluates recommended data types and volumes required to establish a robust de novo genome assembly pipeline for population-level pangenome projects, extensively examining performance between ONT's Duplex and PacBio HiFi datasets in the context of achieving high-quality phased genomes with enhanced contiguity and completeness. The results show that achieving chromosome-level haplotype-resolved assembly requires 20 × high-quality long reads such as PacBio HiFi or ONT Duplex, combined with 15-20 × of ultra-long ONT per haplotype and 10 × of long-range data such as Omni-C or Hi-C. High-quality long reads from both platforms yield assemblies with comparable contiguity, with HiFi excelling in phasing accuracies, while Duplex generates more T2T contigs.
Our study provides insights into optimal data types and volumes for robust de novo genome assembly in population-level pangenome projects. Reassessing the recommended data types and volumes in this study and aligning them with practical economic limitations are vital to the pangenome research community, contributing to their efforts and pushing genomic studies with broader impacts.
太平洋生物科学公司(PacBio)和牛津纳米孔技术公司(ONT)的长读长技术通过提供如HiFi、双链和超长ONT等多种数据类型,改变了基因组学研究。尽管最近在使用长读长技术实现单倍型定相的无间隙基因组组装方面取得了进展,但对于遗传多样性的代表性仍存在担忧,这促使了泛基因组参考的发展。然而,泛基因组研究在为每个组装基因组考虑数据类型、数量和成本方面面临挑战,同时还要努力保持敏感性。缺乏关于最佳数据选择的全面指导加剧了这些挑战。
我们的研究评估了为群体水平的泛基因组项目建立稳健的从头基因组组装流程所需的推荐数据类型和数量,在实现具有更高连续性和完整性的高质量定相基因组的背景下,广泛研究了ONT的双链和PacBio HiFi数据集之间的性能。结果表明,要实现染色体水平的单倍型解析组装,需要20倍的高质量长读长,如PacBio HiFi或ONT双链,再加上每个单倍型15 - 20倍的超长ONT以及10倍的长程数据,如全基因组染色质构象捕获技术(Omni-C)或高通量染色体构象捕获技术(Hi-C)。来自两个平台的高质量长读长产生的组装具有可比的连续性,HiFi在定相准确性方面表现出色,而双链则产生更多的端粒到端粒(T2T)连续片段。
我们的研究为群体水平的泛基因组项目中稳健的从头基因组组装的最佳数据类型和数量提供了见解。重新评估本研究中推荐的数据类型和数量,并使其与实际经济限制相匹配,对泛基因组研究社区至关重要,有助于他们的工作并推动具有更广泛影响的基因组研究。