State Key Laboratory of Silkworm Genome Biology, Biological Science Research Center, Southwest University, Chongqing 400715, China.
Chongqing Key Laboratory of Sericulture Science, Chongqing Engineering and Technology Research Center for Novel Silk Materials, Chongqing 400715, China.
Int J Mol Sci. 2022 Dec 30;24(1):649. doi: 10.3390/ijms24010649.
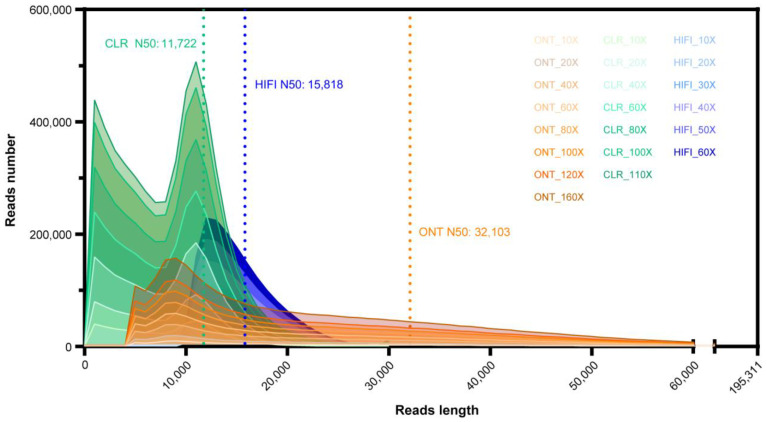
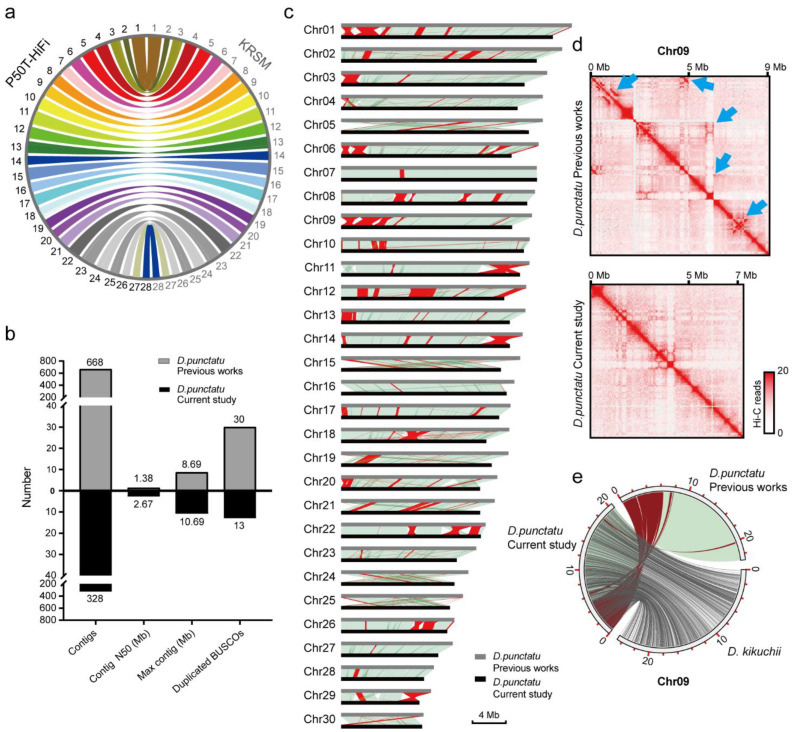
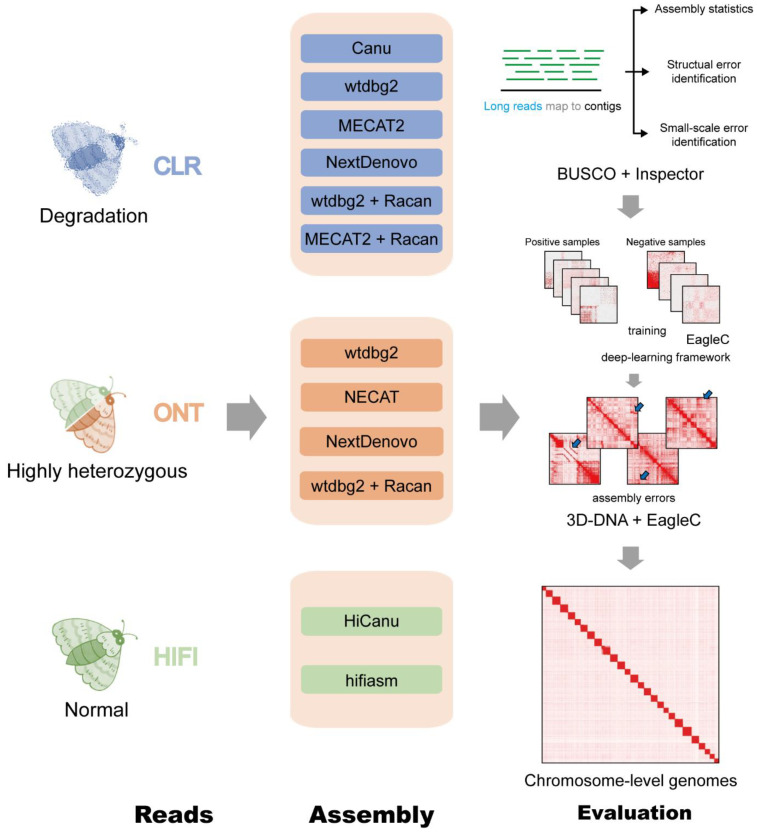
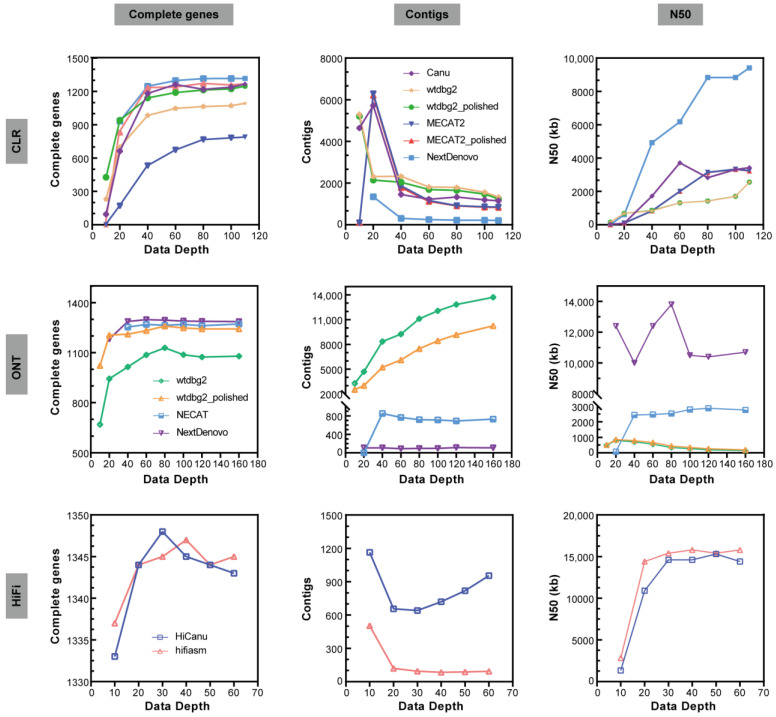
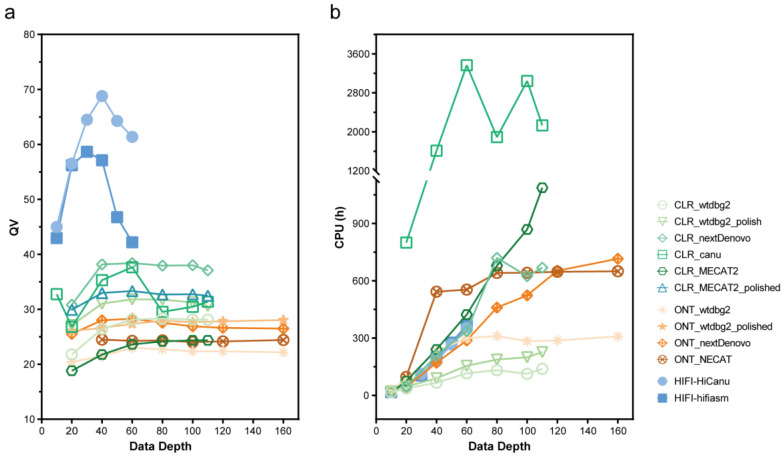
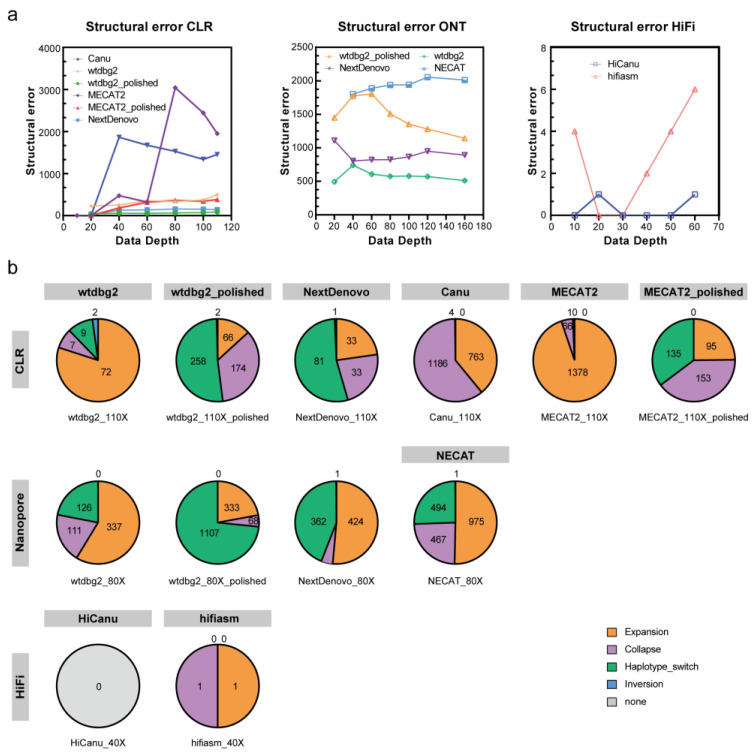
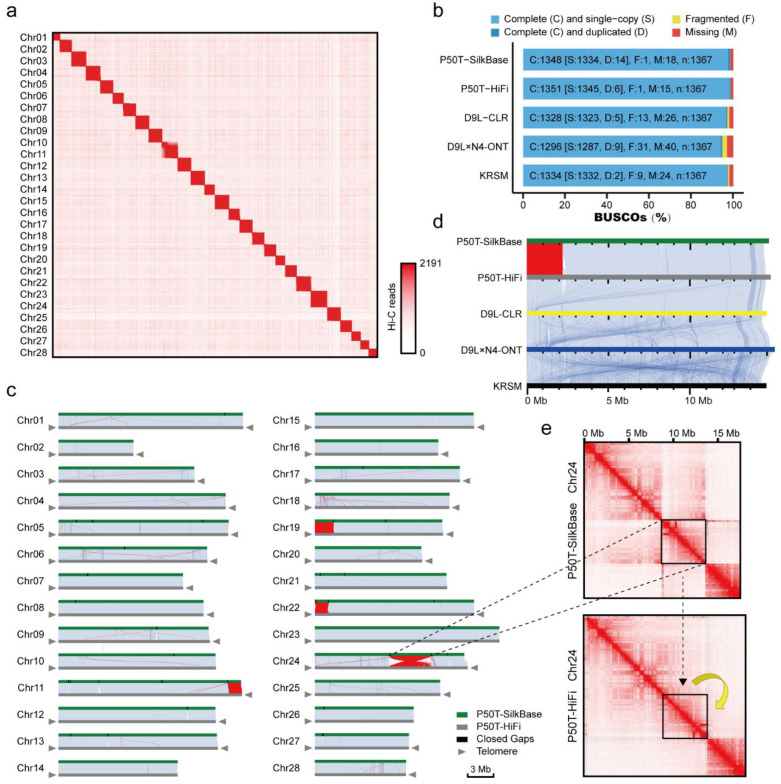
Lepidopteran species are mostly pests, causing serious annual economic losses. High-quality genome sequencing and assembly uncover the genetic foundation of pest occurrence and provide guidance for pest control measures. Long-read sequencing technology and assembly algorithm advances have improved the ability to timeously produce high-quality genomes. Lepidoptera includes a wide variety of insects with high genetic diversity and heterozygosity. Therefore, the selection of an appropriate sequencing and assembly strategy to obtain high-quality genomic information is urgently needed. This research used silkworm as a model to test genome sequencing and assembly through high-coverage datasets by de novo assemblies. We report the first nearly complete telomere-to-telomere reference genome of silkworm (P50T strain) produced by Pacific Biosciences (PacBio) HiFi sequencing, and highly contiguous and complete genome assemblies of two other silkworm strains by Oxford Nanopore Technologies (ONT) or PacBio continuous long-reads (CLR) that were unrepresented in the public database. Assembly quality was evaluated by use of BUSCO, Inspector, and EagleC. It is necessary to choose an appropriate assembler for draft genome construction, especially for low-depth datasets. For PacBio CLR and ONT sequencing, NextDenovo is superior. For PacBio HiFi sequencing, hifiasm is better. Quality assessment is essential for genome assembly and can provide better and more accurate results. For chromosome-level high-quality genome construction, we recommend using 3D-DNA with EagleC evaluation. Our study references how to obtain and evaluate high-quality genome assemblies, and is a resource for biological control, comparative genomics, and evolutionary studies of Lepidopteran pests and related species.
鳞翅目物种大多是害虫,每年造成严重的经济损失。高质量的基因组测序和组装揭示了害虫发生的遗传基础,并为害虫防治措施提供了指导。长读测序技术和组装算法的进步提高了及时产生高质量基因组的能力。鳞翅目包括具有高度遗传多样性和杂合性的多种昆虫。因此,迫切需要选择适当的测序和组装策略来获取高质量的基因组信息。本研究以家蚕为模型,通过从头组装对高覆盖数据集进行基因组测序和组装测试。我们报告了第一个通过太平洋生物科学(PacBio)HiFi 测序生成的家蚕(P50T 品系)近乎完整的端粒到端粒参考基因组,以及两个其他家蚕品系的高度连续和完整基因组组装,这些组装品系在公共数据库中没有代表。使用 BUSCO、Inspector 和 EagleC 评估了组装质量。有必要选择合适的组装器来构建草图基因组,特别是对于低深度数据集。对于 PacBio CLR 和 ONT 测序,NextDenovo 更优。对于 PacBio HiFi 测序,hifiasm 更好。基因组组装的质量评估是必要的,可以提供更好和更准确的结果。对于染色体水平的高质量基因组构建,我们建议使用 EagleC 评估的 3D-DNA。我们的研究参考了如何获得和评估高质量的基因组组装,是鳞翅目害虫和相关物种的生物防治、比较基因组学和进化研究的资源。