Lin Ling, Guo Chunmao, Jin Hanna, Huang Haixiong, Luo Fan, Wang Ying, Li Dongqi, Zhang Yuanxin, Xu Yuqian, Zhu Chanyan, Zeng Fengshan, He Huahua, Chen Jie, Zhang Wei, Yu Wenlin
Department of Neurology, Huizhou Hospital of Guangzhou University of Chinese Medicine (Huizhou Hospital of Traditional Chinese Medicine), Huizhou, Guangdong, China.
Clinical Laboratory, Huizhou Hospital of Guangzhou University of Chinese Medicine (Huizhou Hospital of Traditional Chinese Medicine), Huizhou, Guangdong, China.
Front Neurol. 2024 Dec 4;15:1475582. doi: 10.3389/fneur.2024.1475582. eCollection 2024.
Ischemic stroke (IS) is a significant global health issue, causing high rates of morbidity, mortality, and disability. Since conventional Diagnosis methods for IS have several shortcomings. It is critical to create new Diagnosis models in order to enhance existing Diagnosis approaches.
We utilized gene expression data from the Gene Expression Omnibus (GEO) databases GSE16561 and GSE22255 to identify differentially expressed genes (DEGs) associated with IS. DEGs analysis using the Limma package, as well as GO and KEGG enrichment analyses, were performed. Furthermore, PPI networks were constructed using DEGs from the String database, and Random Forest models were utilized to screen key DEGs. Additionally, an artificial neural network model was developed for IS classification. Use the GSE58294 dataset to evaluate the effectiveness of the scoring model on healthy controls and ischemic stroke samples. The effectiveness of the scoring model was evaluated through AUC analysis, and CIBERSORT analysis was conducted to estimate the immune landscape and explore the correlation between gene expression and immune cell infiltration.
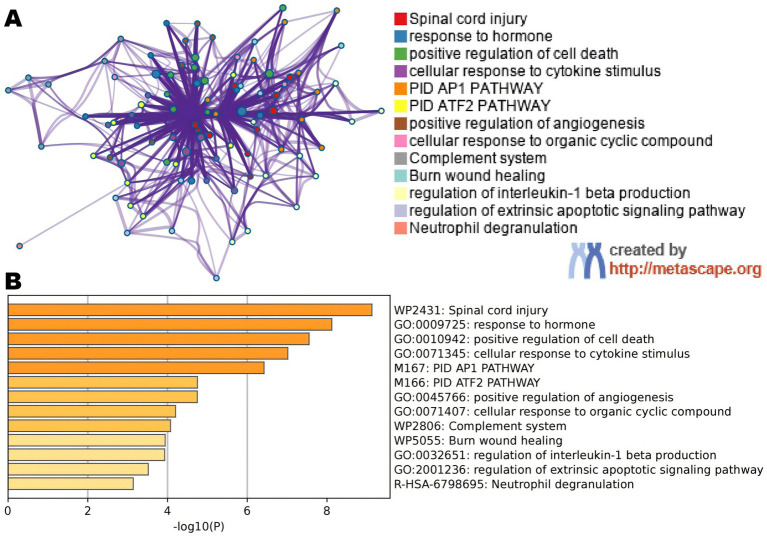
A total of 26 significant DEGs associated with IS were identified. Metascape analysis revealed enriched biological processes and pathways related to IS. 10 key DEGs (ARG1, DUSP1, F13A1, NFIL3, CCR7, ADM, PTGS2, ID3, FAIM3, HLA-DQB1) were selected using Random Forest and artificial neural network models. The area under the ROC curve (AUC) for the IS classification model was found to be near 1, indicating its high accuracy. Additionally, the analysis of the immune landscape demonstrated elevated immune-related networks in IS patients compared to healthy controls.
The study uncovers the involvement of specific genes and immune cells in the pathogenesis of IS, suggesting their importance in understanding and potentially targeting the disease.
缺血性中风(IS)是一个重大的全球健康问题,导致高发病率、死亡率和残疾率。由于IS的传统诊断方法存在若干缺点,创建新的诊断模型以改进现有诊断方法至关重要。
我们利用来自基因表达综合数据库(GEO)的GSE16561和GSE22255基因表达数据来识别与IS相关的差异表达基因(DEG)。使用Limma软件包进行DEG分析以及GO和KEGG富集分析。此外,利用来自String数据库的DEG构建蛋白质-蛋白质相互作用(PPI)网络,并使用随机森林模型筛选关键DEG。另外,开发了一种用于IS分类的人工神经网络模型。使用GSE58294数据集评估评分模型对健康对照和缺血性中风样本的有效性。通过曲线下面积(AUC)分析评估评分模型的有效性,并进行CIBERSORT分析以估计免疫格局并探索基因表达与免疫细胞浸润之间的相关性。
共鉴定出26个与IS相关的显著DEG。Metascape分析揭示了与IS相关的丰富生物过程和途径。使用随机森林和人工神经网络模型选择了10个关键DEG(ARG1、DUSP1、Fgb、NFIL3、CCR7、ADM、PTGS2、ID3、FAIM3、HLA-DQB1)。发现IS分类模型的ROC曲线下面积(AUC)接近1,表明其具有高准确性。此外,免疫格局分析表明,与健康对照相比,IS患者的免疫相关网络升高。
该研究揭示了特定基因和免疫细胞在IS发病机制中的作用,表明它们在理解和潜在靶向该疾病方面的重要性。