Li Chenhe, Zhang Lei, Peng Zhibang, Li Xia, Liu Zhao, Lu Te, Kang Xiangyang, Yang Jun
State Key Laboratory of Tree Genetics and Breeding, National Engineering Research Center of Tree Breeding and Ecological Restoration, Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental Plants of Ministry of Education, College of Biological Sciences and Technology, Beijing Forestry University, Beijing, 100083, China.
Guangxi Dongmen Forest Farm, Chongzuo, 532199, China.
BMC Plant Biol. 2024 Dec 23;24(1):1240. doi: 10.1186/s12870-024-05970-0.
Eucalyptus grandis, which was first comprehensively and systematically introduced to China in the 1980s, is one of the most important fast-growing tree species in the forestry industry. However, to date, no core collection has been selected from the germplasm resources of E. grandis based on growth and genetic relationship analysis.
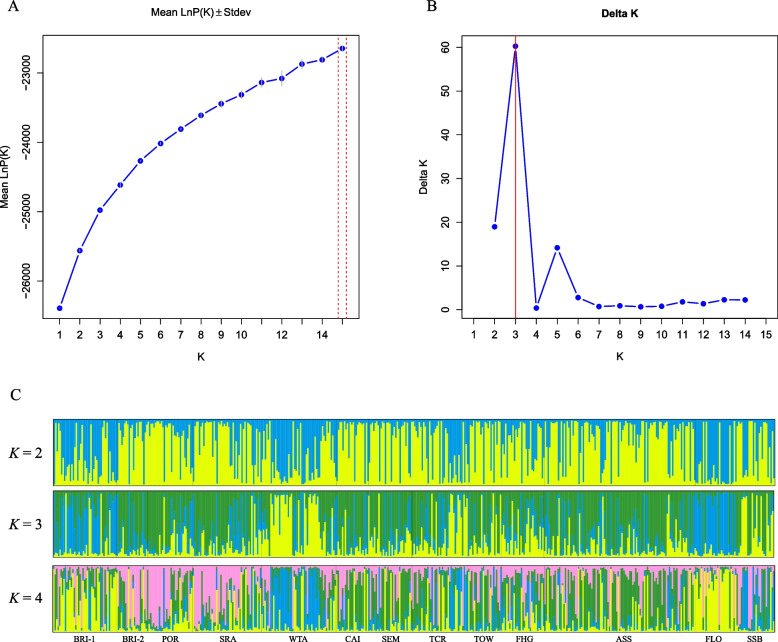
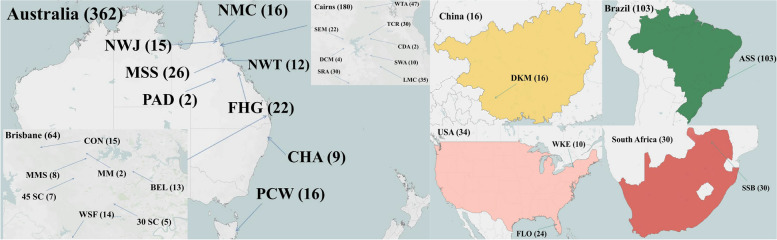
In the present study, 545 individuals of E. grandis collected from 28 populations across 5 countries were selected for genetic diversity analysis using 16 selected SSR markers. The polymorphism information content (PIC) was employed to assess genetic diversity, yielding a mean value of 0.707. Genetic structure analysis was conducted on 492 individuals from 13 combined populations, revealing three clusters as the most suitable number. Principal coordinate analysis (PCoA) demonstrated that the populations were divided into three major clusters. Additionally, the analysis of molecular variance (AMOVA) indicated that the majority of variation occurred within populations.
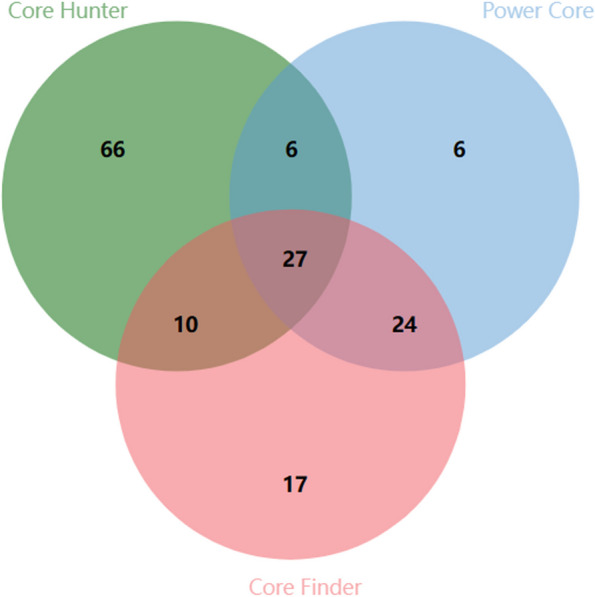
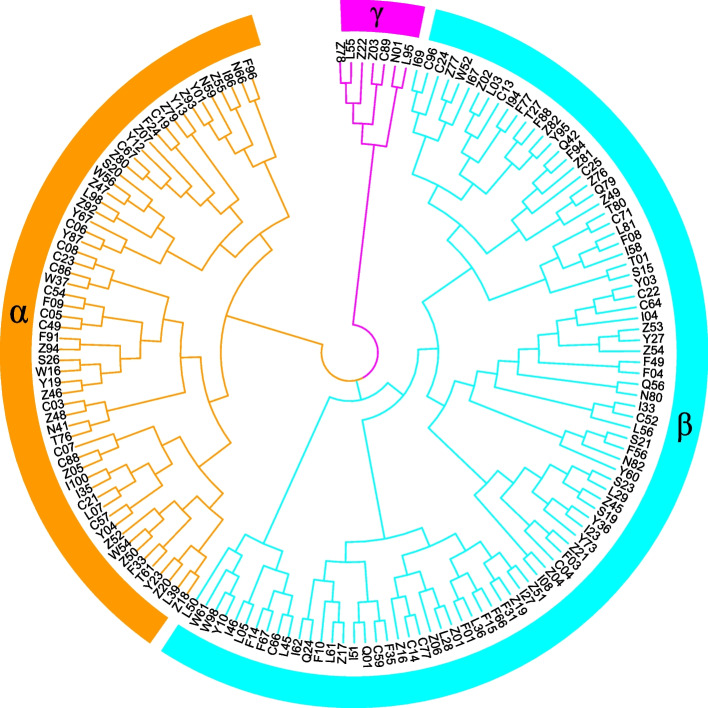
Based on the criteria for screening the core collection, we constructed a population consisting of 158 individuals and created unique fingerprinting codes. These results provide a crucial theoretical foundation for the protection and utilization of germplasm resources of E. grandis in China, which will be helpful in the selection of genetically distant parents for future multigenerational hybridization programs.
巨桉于20世纪80年代首次被全面系统地引入中国,是林业中最重要的速生树种之一。然而,迄今为止,尚未基于生长和遗传关系分析从巨桉种质资源中筛选出核心种质库。
在本研究中,从5个国家的28个种群中收集了545株巨桉个体,使用16个选定的SSR标记进行遗传多样性分析。采用多态性信息含量(PIC)评估遗传多样性,平均值为0.707。对来自13个合并种群的492个个体进行遗传结构分析,结果显示最合适的聚类数为3个。主坐标分析(PCoA)表明种群被分为3个主要聚类。此外,分子方差分析(AMOVA)表明大部分变异发生在种群内部。
基于核心种质库的筛选标准,我们构建了一个由158个个体组成的种群,并创建了独特的指纹编码。这些结果为中国巨桉种质资源的保护和利用提供了关键的理论基础,这将有助于为未来的多代杂交计划选择遗传距离较远的亲本。