Chauhan Siddharth M, Ardalani Omid, Hyun Jason C, Monk Jonathan M, Phaneuf Patrick V, Palsson Bernhard O
Department of Bioengineering, University of California, San Diego, La Jolla, California, USA.
Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet, Kongens, Lyngby, Denmark.
mSphere. 2025 Jan 28;10(1):e0053224. doi: 10.1128/msphere.00532-24. Epub 2024 Dec 31.
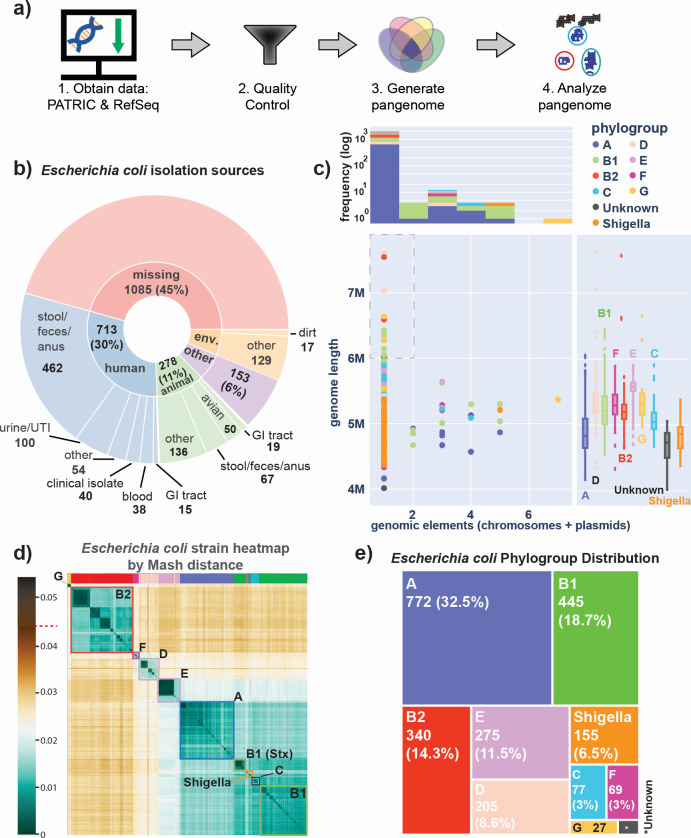
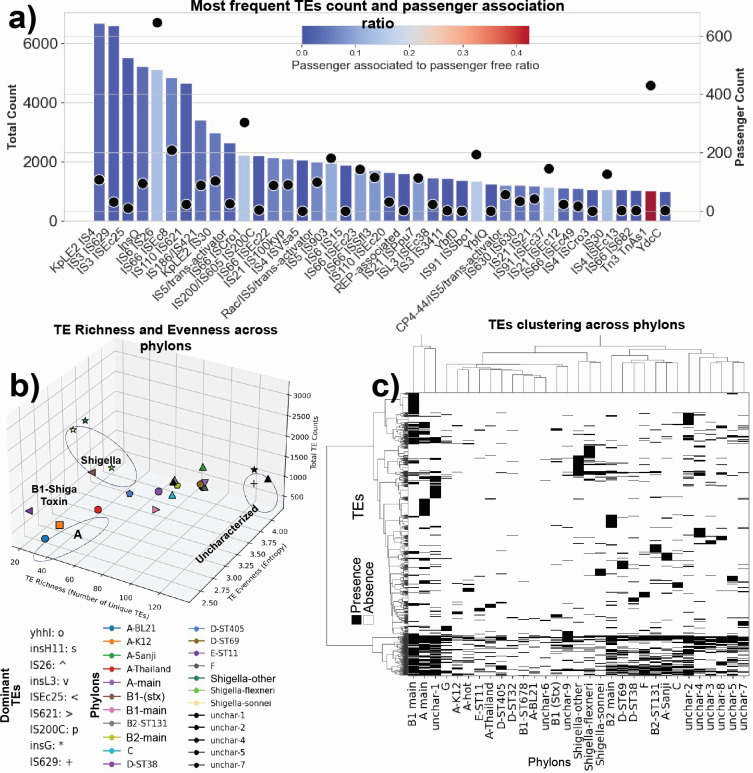
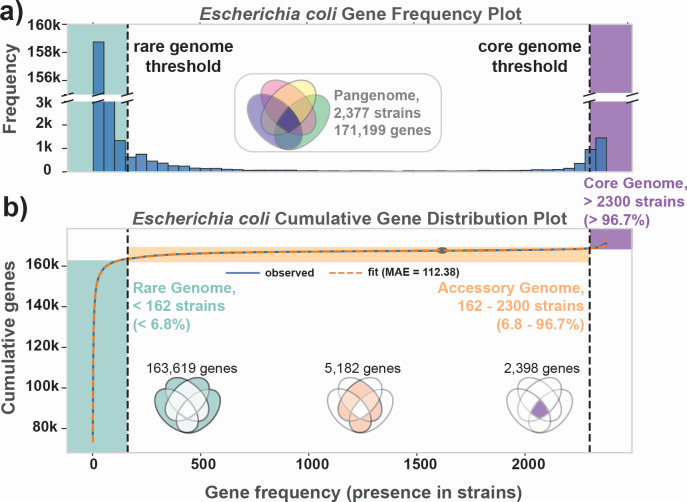
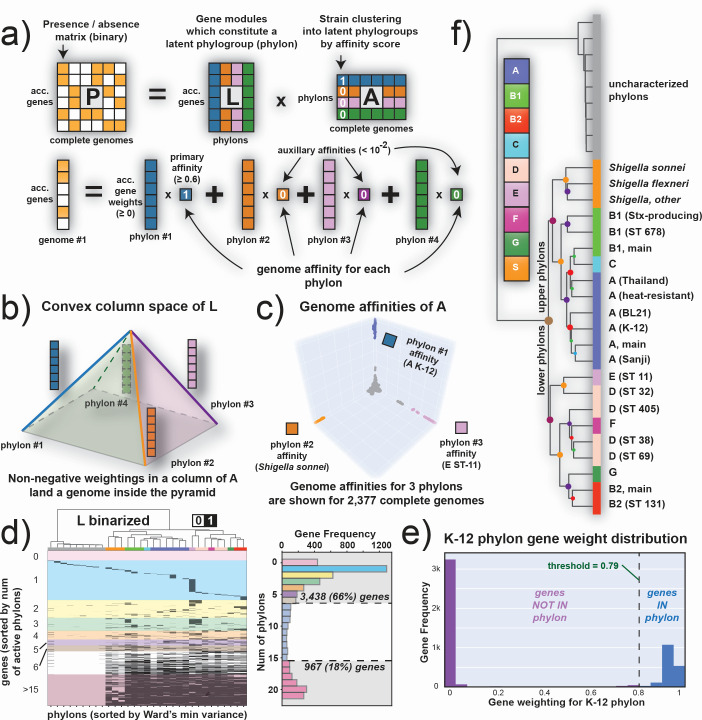
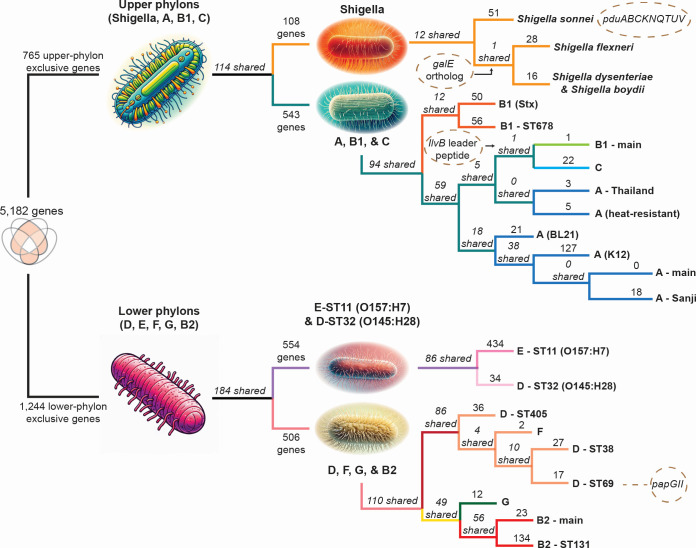
Thousands of complete genome sequences for strains of a species that are now available enable the advancement of pangenome analytics to a new level of sophistication. We collected 2,377 publicly available complete genomes of for detailed pangenome analysis. The core genome and accessory genomes consisted of 2,398 and 5,182 genes, respectively. We developed a machine learning approach to define the accessory genes characterizing the major phylogroups of plus : A, B1, B2, C, D, E, F, G, and . The analysis resulted in a detailed structure of the genetic basis of the phylogroups' differential traits. This pangenome structure was largely consistent with a housekeeping-gene-based MLST distribution, sequence-based Mash distance, and the Clermont quadruplex classification. The rare genome (consisting of genes found in <6.8% of all strains) consisted of 163,619 genes, about 79% of which represented variations of 315 underlying transposon elements. This analysis generated a mathematical definition of the genetic basis for a species.
The comprehensive analysis of the pangenome of presented in this study marks a significant advancement in understanding bacterial genetic diversity. By employing machine learning techniques to analyze 2,377 complete genomes, the study provides a detailed mapping of core, accessory, and rare genes. This approach reveals the genetic basis for differential traits across phylogroups, offering insights into pathogenicity, antibiotic resistance, and evolutionary adaptations. The findings enhance the potential for genome-based diagnostics and pave the way for future studies aimed at achieving a global genetic definition of bacterial phylogeny.
现在已有一个物种菌株的数千个完整基因组序列,这使得泛基因组分析能够提升到一个新的复杂程度。我们收集了2377个公开可用的完整基因组用于详细的泛基因组分析。核心基因组和辅助基因组分别由2398个和5182个基因组成。我们开发了一种机器学习方法来定义表征该物种加上某些其他分类群(A、B1、B2、C、D、E、F、G以及某些其他分类群)主要系统发育群的辅助基因。分析得出了系统发育群差异特征遗传基础的详细结构。这种泛基因组结构在很大程度上与基于管家基因的多位点序列分型分布、基于序列的Mash距离以及克莱蒙特四重分类法一致。稀有基因组(由在所有菌株中<6.8%的菌株中发现的基因组成)由163619个基因组成,其中约79%代表315个潜在转座子元件的变异。该分析生成了一个物种遗传基础的数学定义。
本研究中对该物种泛基因组的全面分析标志着在理解细菌遗传多样性方面取得了重大进展。通过运用机器学习技术分析2377个完整的该物种基因组,该研究提供了核心、辅助和稀有基因的详细图谱。这种方法揭示了不同系统发育群差异特征的遗传基础,为致病性、抗生素抗性和进化适应性提供了见解。这些发现增强了基于基因组的诊断潜力,并为旨在实现细菌系统发育全球遗传定义的未来研究铺平了道路。