Khadka Mukesh, Sah Manoj, Chaudhary Raju, Sahani Suresh Kumar, Sahani Kameshwar, Pandey Binay Kumar, Pandey Digvijay
Department of Physics, St.Xavier College, Maitighar, Kathmandu, Nepal.
Department of Science and Technology, Rajarshi Janak University, Janakpurdham, Nepal.
Sci Rep. 2025 Jan 2;15(1):208. doi: 10.1038/s41598-024-81633-2.
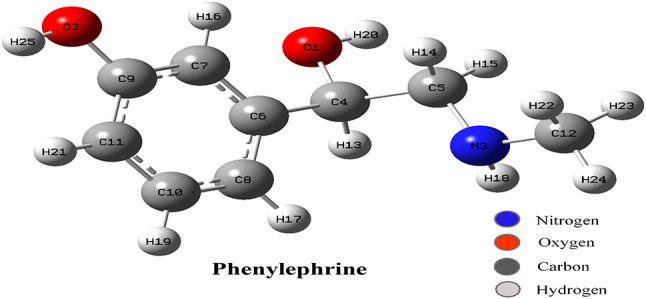
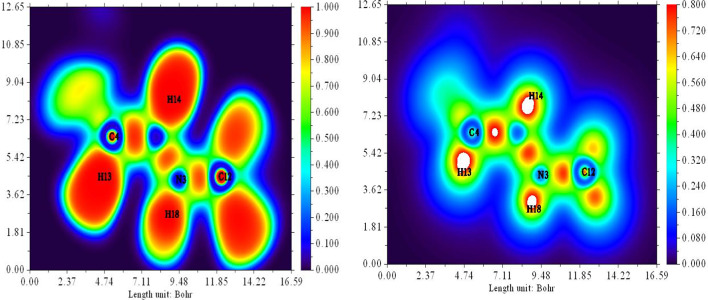
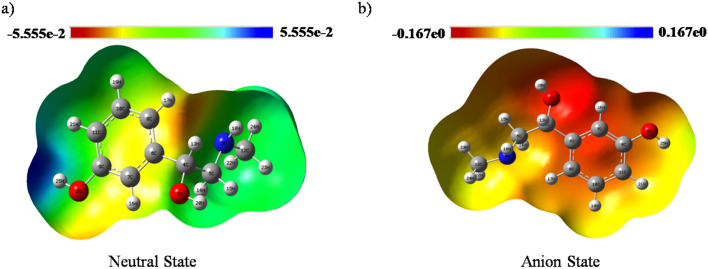
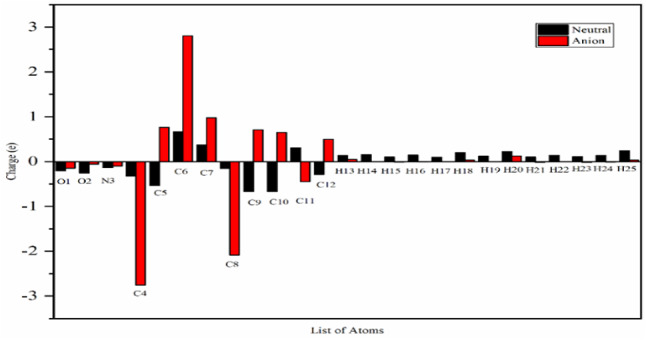
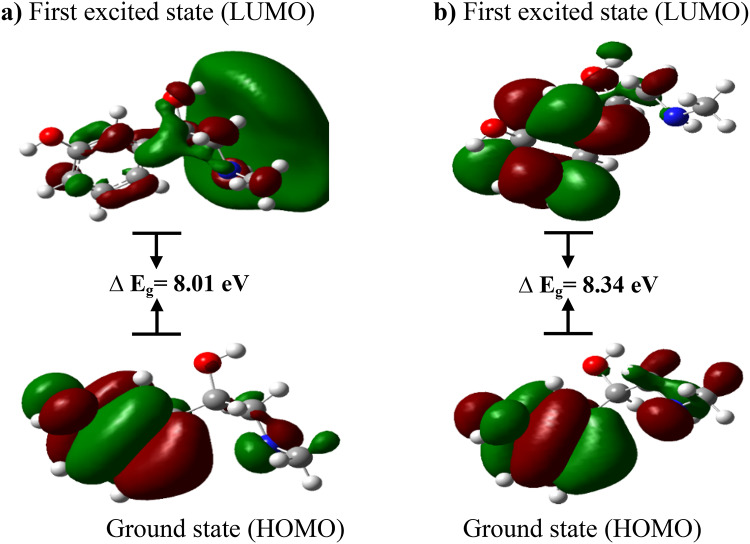
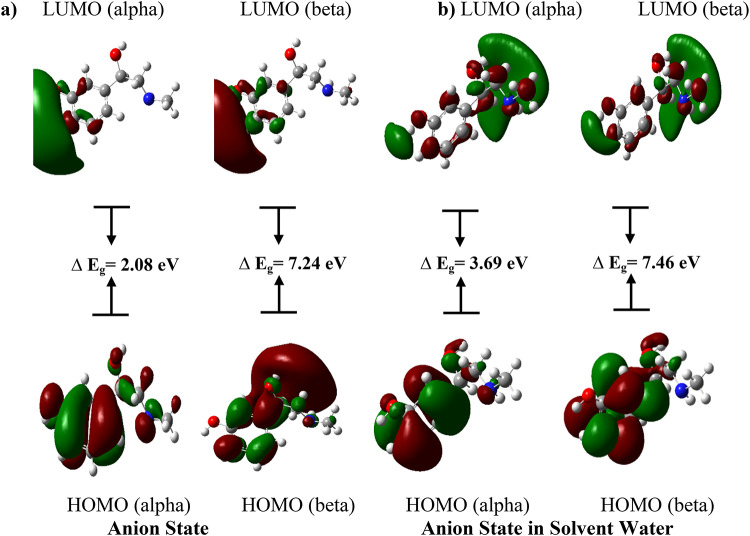
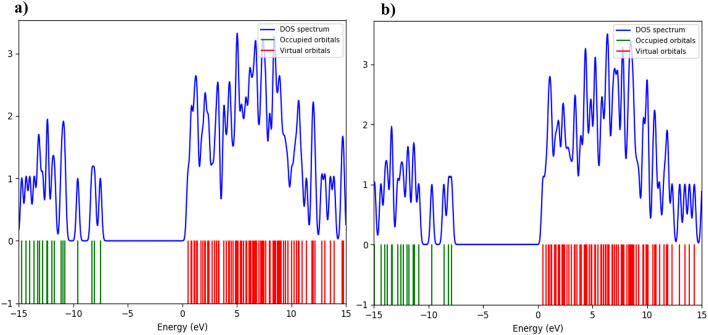
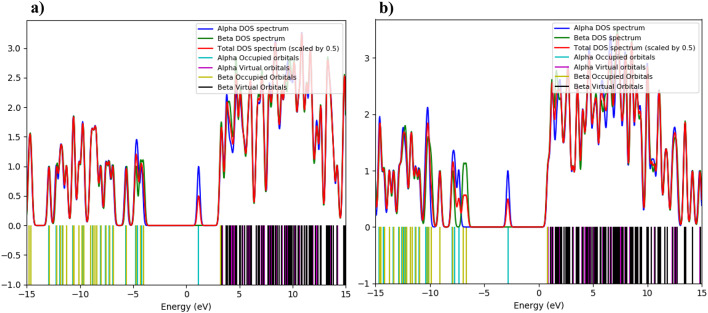
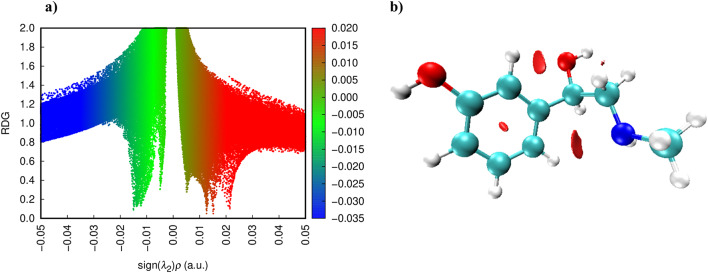
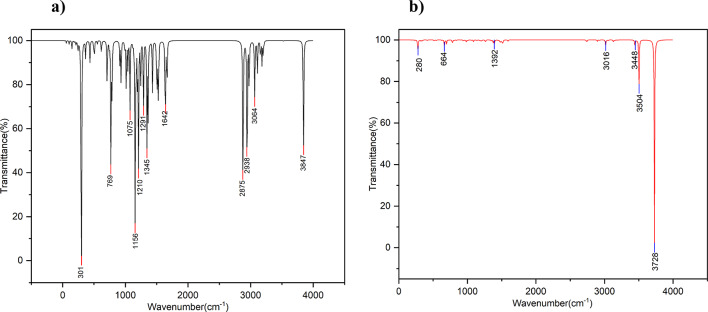
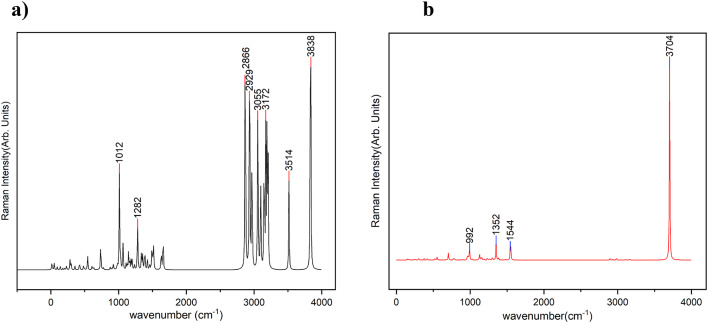
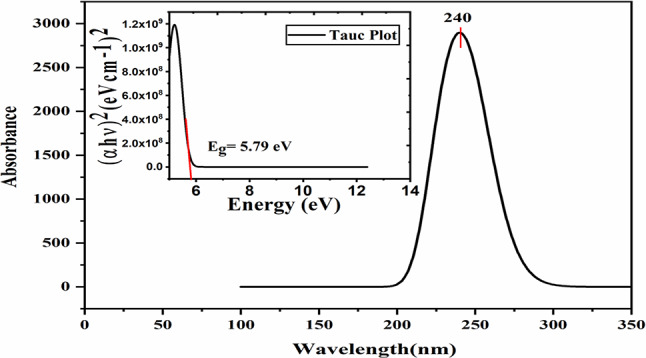
In this work, Density Functional Theory (DFT) on Gaussian 09 W software was utilized to investigate the phenylephrine (PE) molecule (C9H13NO2). Firstly, the optimized structure of the PE molecule was obtained using B3LYP/6-311 + G (d, p) and CAM-B3LYP/6-311 + G (d, p) basis sets. The electron charge density is shown in Mulliken atomic charge as a bar chart and also as a color-filled map in Molecular Electrostatic Potential (MEP). Using these properties, the possibility of different charge transfers occurring within the molecule was evaluated. The calculated values of the energy gap from HOMO-LUMO mapping, illustrated in Frontier Molecular Orbitals (FMO) and Density of State (DOS), were found to be similar for both the neutral and anion states in the gaseous and water solvent phases. Both the global and local reactivity were studied to understand the reactivity of the PE molecule. Using the thermodynamic parameters, the thermochemical property of the title molecule was understood. Non-covalent interaction was studied to understand the Van der Waals interactions, hydrogen bonds, and steric repulsion in the title molecule. Natural Bond Orbital (NBO) Analysis was performed to understand the strongest stabilization interaction. In the vibrational analysis, Total Electron Density (TED) assignments were done in the intense region where the frequency of the title molecule was shifted distinctly. For vibrational spectroscopy, FT-IR and Raman spectra in the neutral and anion states were plotted and compared. Using the TD-DFT technique, the UV-Vis spectra along with Tauc's plot were studied. Finally, topological analysis, electron localized function (ELF), and localized orbital locator (LOL) were performed in the PE molecule.
在这项工作中,利用高斯09 W软件上的密度泛函理论(DFT)来研究去氧肾上腺素(PE)分子(C9H13NO2)。首先,使用B3LYP/6-311+G(d,p)和CAM-B3LYP/6-311+G(d,p)基组获得了PE分子的优化结构。电子电荷密度以穆利肯原子电荷的柱状图形式显示,也以分子静电势(MEP)中的彩色填充图形式显示。利用这些性质,评估了分子内不同电荷转移发生的可能性。在前沿分子轨道(FMO)和态密度(DOS)中所示的从HOMO-LUMO映射计算得到的能隙值,在气态和水溶剂相中的中性和阴离子态下均相似。研究了全局和局部反应性以了解PE分子的反应性。利用热力学参数,了解了标题分子的热化学性质。研究了非共价相互作用以了解标题分子中的范德华相互作用、氢键和空间排斥。进行了自然键轨道(NBO)分析以了解最强的稳定相互作用。在振动分析中,在标题分子频率明显偏移的强区域进行了总电子密度(TED)归属。对于振动光谱,绘制并比较了中性和阴离子态下的傅里叶变换红外光谱(FT-IR)和拉曼光谱。使用TD-DFT技术,研究了紫外可见光谱以及陶氏图。最后,在PE分子中进行了拓扑分析、电子定域函数(ELF)和定域轨道定位器(LOL)分析。