Zhang Xiang, Ling Tianze, Jin Zhi, Xu Sheng, Gao Zhiqiang, Sun Boyan, Qiu Zijie, Wei Jiaqi, Dong Nanqing, Wang Guangshuai, Wang Guibin, Li Leyuan, Abdul-Mageed Muhammad, Lakshmanan Laks V S, He Fuchu, Ouyang Wanli, Chang Cheng, Sun Siqi
Shanghai Artificial Intelligence Laboratory, Shanghai, China.
University of British Columbia, Vancouver, BC, Canada.
Nat Commun. 2025 Jan 2;16(1):267. doi: 10.1038/s41467-024-55021-3.
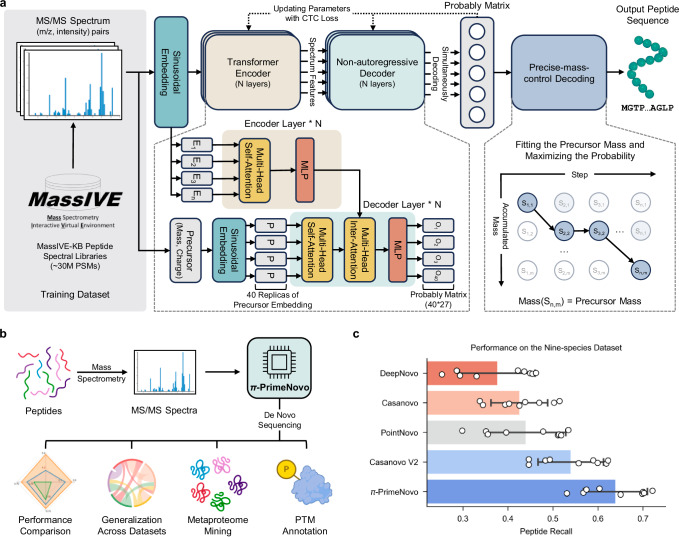
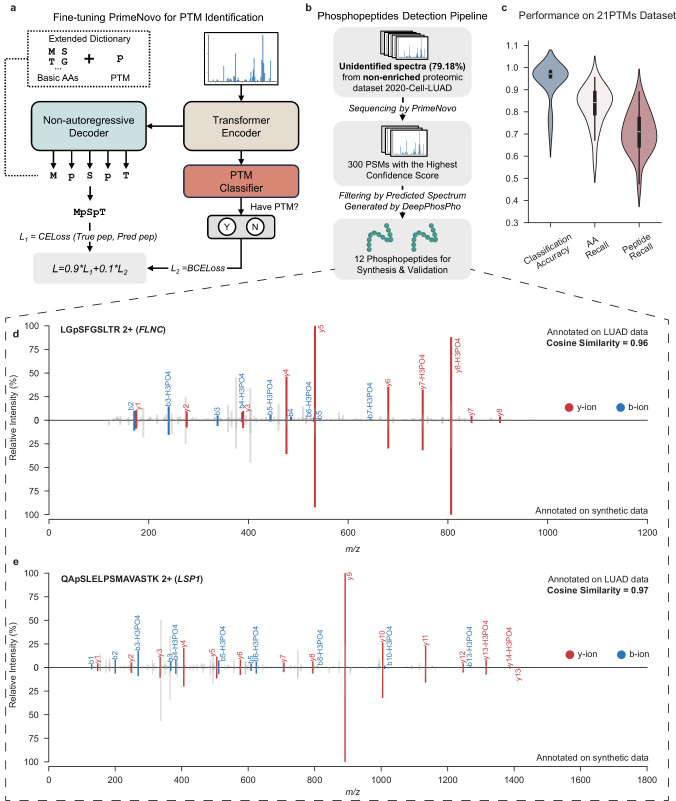
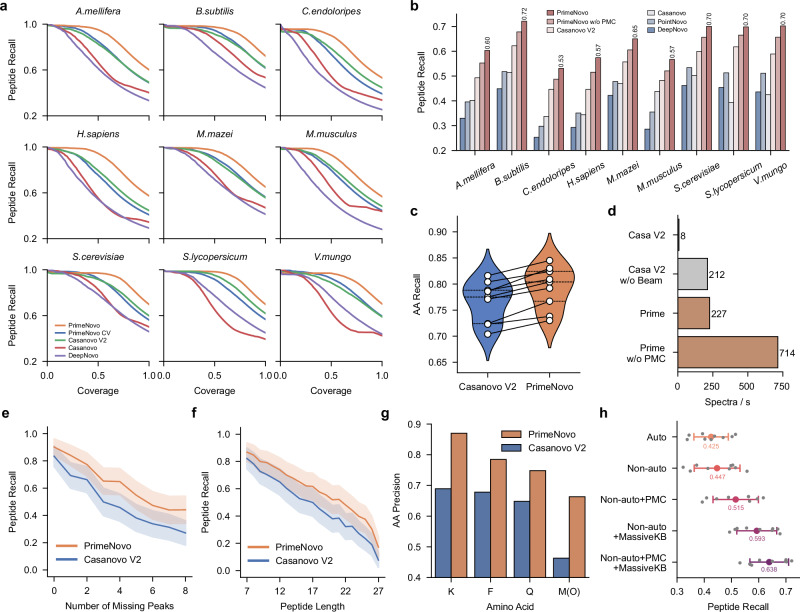
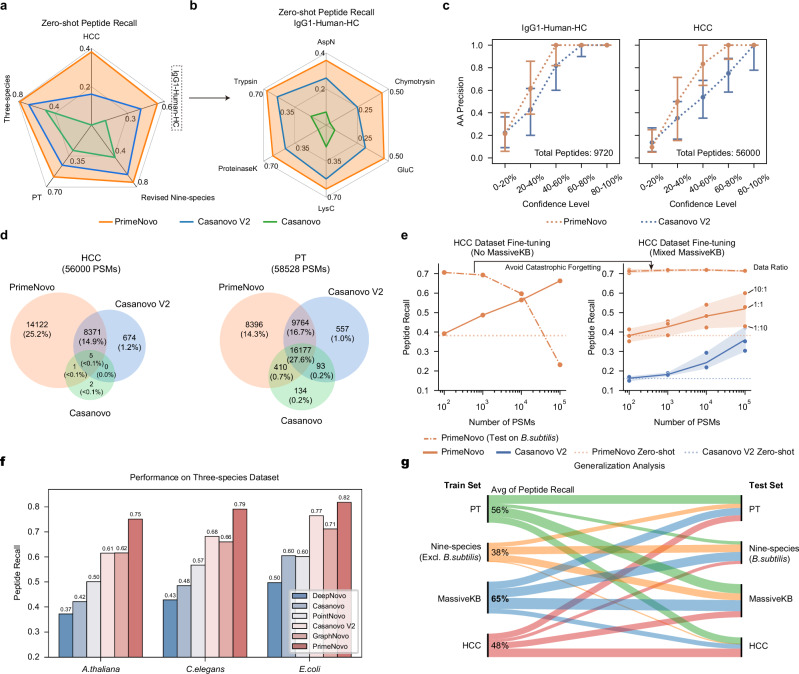
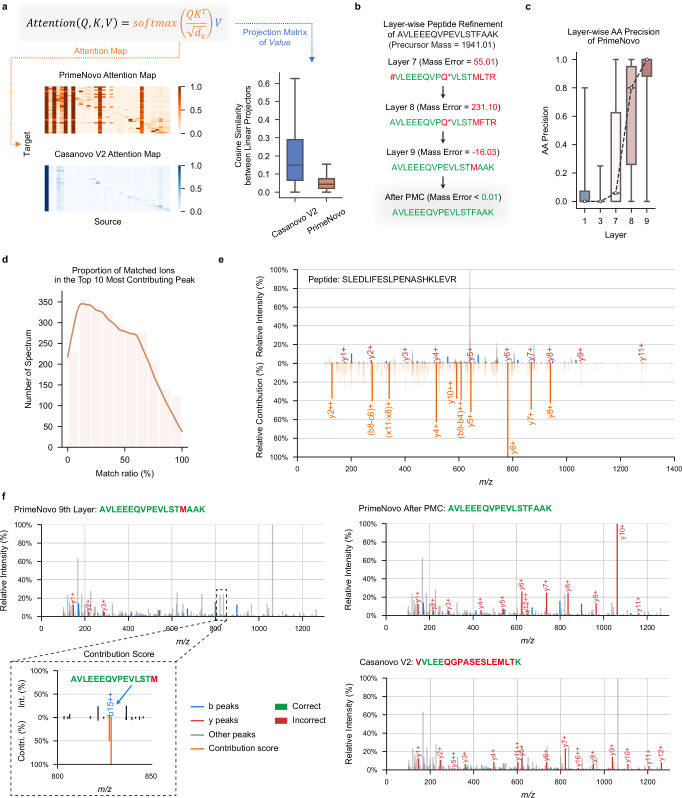
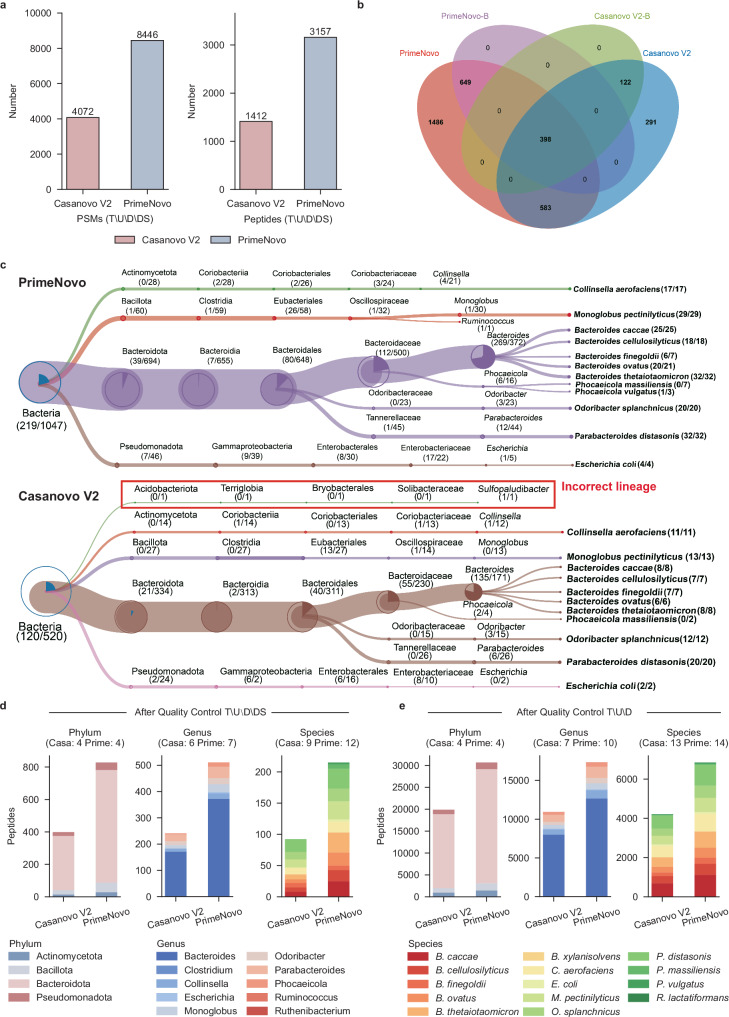
Peptide sequencing via tandem mass spectrometry (MS/MS) is essential in proteomics. Unlike traditional database searches, deep learning excels at de novo peptide sequencing, even for peptides missing from existing databases. Current deep learning models often rely on autoregressive generation, which suffers from error accumulation and slow inference speeds. In this work, we introduce π-PrimeNovo, a non-autoregressive Transformer-based model for peptide sequencing. With our architecture design and a CUDA-enhanced decoding module for precise mass control, π-PrimeNovo achieves significantly higher accuracy and up to 89x faster inference than state-of-the-art methods, making it ideal for large-scale applications like metaproteomics. Additionally, it excels in phosphopeptide mining and detecting low-abundance post-translational modifications (PTMs), marking a substantial advance in peptide sequencing with broad potential in biological research.
通过串联质谱(MS/MS)进行肽段测序在蛋白质组学中至关重要。与传统的数据库搜索不同,深度学习在从头肽段测序方面表现出色,即使对于现有数据库中缺失的肽段也是如此。当前的深度学习模型通常依赖自回归生成,这存在误差累积和推理速度慢的问题。在这项工作中,我们引入了π-PrimeNovo,这是一种基于非自回归Transformer的肽段测序模型。通过我们的架构设计和用于精确质量控制的CUDA增强解码模块,π-PrimeNovo实现了比现有方法显著更高的准确性,推理速度快达89倍,使其成为宏蛋白质组学等大规模应用的理想选择。此外,它在磷酸肽挖掘和检测低丰度翻译后修饰(PTM)方面表现出色,标志着肽段测序取得了重大进展,在生物学研究中具有广泛的潜力。