Robert Koch Institute, MF1, Nordufer 20, 13353 Berlin.
Federal Institute for Materials Research and Testing (BAM), Richard-Willstätter-Straße 11, 12489 Berlin.
Brief Bioinform. 2023 Jan 19;24(1). doi: 10.1093/bib/bbac542.
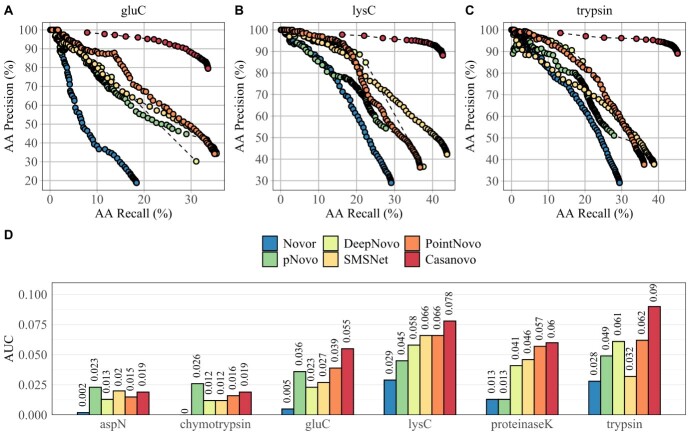
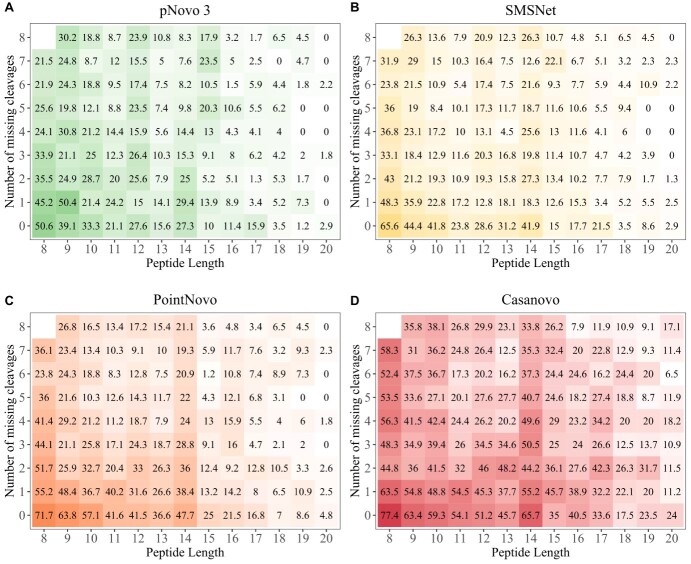
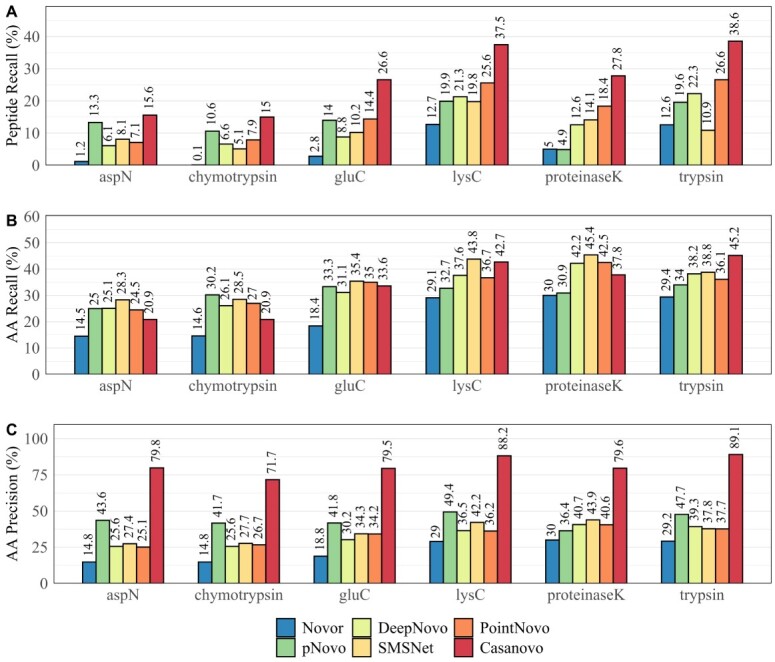
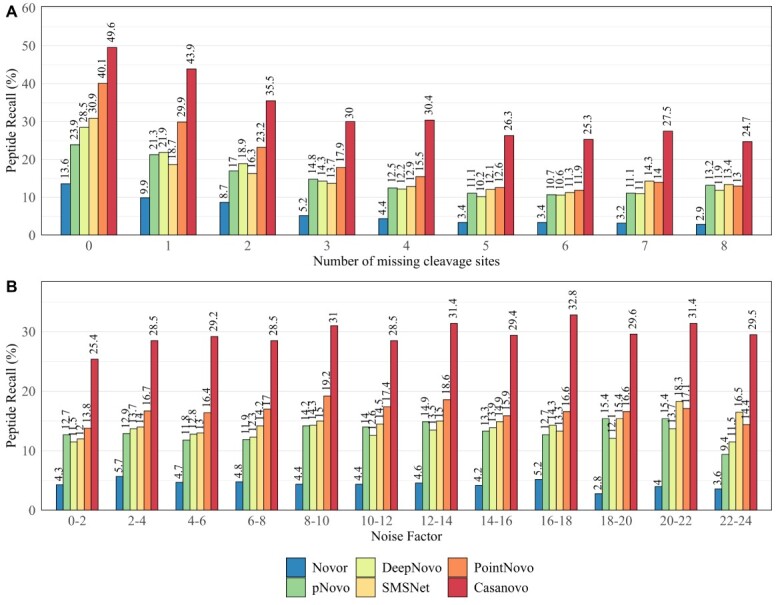
Monoclonal antibodies are biotechnologically produced proteins with various applications in research, therapeutics and diagnostics. Their ability to recognize and bind to specific molecule structures makes them essential research tools and therapeutic agents. Sequence information of antibodies is helpful for understanding antibody-antigen interactions and ensuring their affinity and specificity. De novo protein sequencing based on mass spectrometry is a valuable method to obtain the amino acid sequence of peptides and proteins without a priori knowledge. In this study, we evaluated six recently developed de novo peptide sequencing algorithms (Novor, pNovo 3, DeepNovo, SMSNet, PointNovo and Casanovo), which were not specifically designed for antibody data. We validated their ability to identify and assemble antibody sequences on three multi-enzymatic data sets. The deep learning-based tools Casanovo and PointNovo showed an increased peptide recall across different enzymes and data sets compared with spectrum-graph-based approaches. We evaluated different error types of de novo peptide sequencing tools and their performance for different numbers of missing cleavage sites, noisy spectra and peptides of various lengths. We achieved a sequence coverage of 97.69-99.53% on the light chains of three different antibody data sets using the de Bruijn assembler ALPS and the predictions from Casanovo. However, low sequence coverage and accuracy on the heavy chains demonstrate that complete de novo protein sequencing remains a challenging issue in proteomics that requires improved de novo error correction, alternative digestion strategies and hybrid approaches such as homology search to achieve high accuracy on long protein sequences.
单克隆抗体是生物技术生产的蛋白质,具有广泛的应用于研究、治疗和诊断。它们识别和结合特定分子结构的能力使它们成为重要的研究工具和治疗剂。抗体的序列信息有助于了解抗体-抗原相互作用,并确保其亲和力和特异性。基于质谱的从头蛋白质测序是一种获得肽和蛋白质氨基酸序列的有价值的方法,无需先验知识。在这项研究中,我们评估了六种最近开发的从头多肽测序算法(Novor、pNovo 3、DeepNovo、SMSNet、PointNovo 和 Casanovo),这些算法并非专门为抗体数据设计。我们验证了它们在三个多酶数据集上识别和组装抗体序列的能力。基于深度学习的工具 Casanovo 和 PointNovo 与基于谱图的方法相比,在不同的酶和数据集上提高了肽的召回率。我们评估了从头多肽测序工具的不同错误类型及其在不同数量的缺失切割位点、嘈杂光谱和不同长度肽的性能。我们使用 de Bruijn 组装器 ALPS 和 Casanovo 的预测,在三个不同的抗体数据集的轻链上实现了 97.69-99.53%的序列覆盖率。然而,重链上的低序列覆盖率和准确性表明,完整的从头蛋白质测序仍然是蛋白质组学中的一个具有挑战性的问题,需要改进从头错误纠正、替代消化策略和混合方法,如同源搜索,以实现长蛋白质序列的高精度。