Mahaman Mourtala Issa Zakari, Gouda Arnaud Comlan, Baina Dan-Jimo, Maxwell Nwankwo Innocent Ifeanyi, Adje Charlotte O A, Baragé Moussa, Happiness Oselebe Ogba
Department of Natural Resources Management, National Institute of Agronomic Research of Niger, Niamey, Niger.
Africa Rice Center, M'bé Research Station, Bouaké 01, Côte d'Ivoire.
PLoS One. 2025 Jan 3;20(1):e0312384. doi: 10.1371/journal.pone.0312384. eCollection 2025.
Sweetpotato is a vegetatively propagated crop cultivated worldwide, predominantly in developing countries, valued for its adaptability, short growth cycle, and high productivity per unit land area. In most sub-Saharan African (SSA) countries, it is widely grown by smallholder farmers. Niger, Nigeria, and Benin have a huge diversity of sweetpotato accessions whose potential has not fully been explored to date. Diversity Arrays Technology (DArTseq), a Genotyping by Sequencing (GBS) method, has been developed and enables genotyping with high-density single nucleotide polymorphisms (SNPs) in different crop species. The aim of this study was to assess the genetic diversity and population structure of the West African sweetpotato collection using Diversity Arrays Technology through Genotyping by Sequencing (GBS).
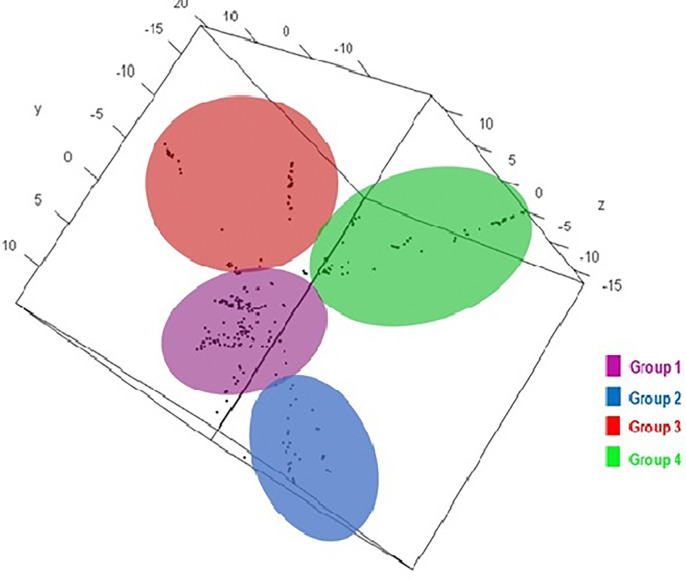
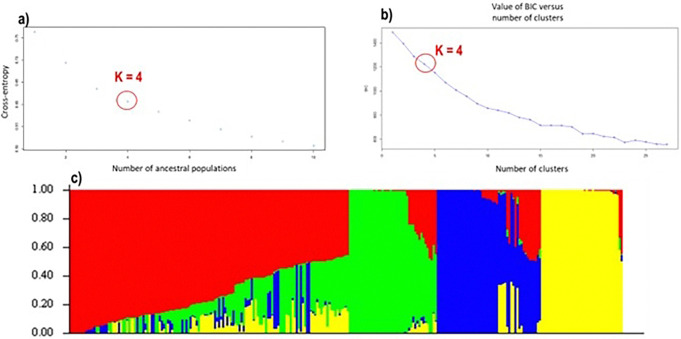
29,523 Diversity Arrays Technology (DArTseq) single nucleotide polymorphism markers were used to genotype 271 sweetpotato accessions. Genetic diversity analysis revealed an average polymorphic information content (PIC) value of 0.39, a minor allele frequency of 0.26, and an observed heterozygosity of 10%. The highest value of polymorphic information content (PIC) (0.41) was observed in chromosomes 4, while the highest proportion of heterozygous (He) (0.18) was observed in chromosomes 11. Molecular diversity revealed high values of polymorphic sites (Ps), theta (θ), and nucleotide diversity (π) with 0.973, 0.158, and 0.086, respectively, which indicated high genetic variation. The pairs of genetic distances revealed a range from 0.08 to 0.47 with an overall average of 0.34. Population structure analysis divided the 271 accessions into four populations (population 1 was characterised by a mixture of accessions from all countries; population 2, mostly comprised of Nigerian breeding lines; population 3 contained exclusively landraces from Benin; and population 4 was composed by only landraces from West African countries) at K = 4, and analysis of molecular variance (AMOVA) based on PhiPT values showed that most of the variation was explained when accessions were categorized based on population structure at K = 4 (25.25%) and based on cluster analysis (19.43%). Genetic distance showed that group 4 (which constituted by landraces of Niger and Benin) was genetically distant (0.428) from groups 2 (formed by 75% of breeding lines of Nigeria), while group 1 was the closest (0.182) to group 2.
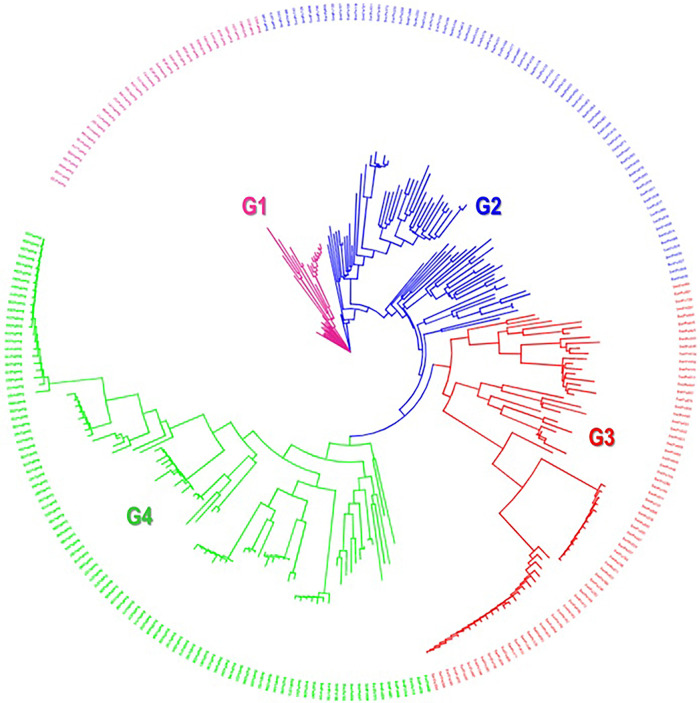
This study employed 7,591 DArTseq-based SNP markers, revealing extensive polymorphism and variation within and between populations. Variability among countries of origin (11.42%) exceeded that based on biological status (9.13%) and storage root flesh colour (7.90%), emphasizing the impact of migration on genetic diversity. Population structure analysis using principal component analysis (PCA), Neighbor-Joining (NJ) tree, and STRUCTURE at K = 4 grouped 271 accessions into distinct clusters, irrespective of their geographic origins, indicating widespread genetic exchange. Group 4, dominated by landraces (95%), showed significant genetic differentiation (Nei's Gst = 0.428) from Group 2, mainly comprising breeding lines, suggesting their potential as heterotic groups for breeding initiatives like HEBS or ABS.
甘薯是一种通过营养繁殖的作物,在全球范围内种植,主要分布在发展中国家,因其适应性强、生长周期短和单位土地面积产量高而受到重视。在大多数撒哈拉以南非洲(SSA)国家,小农户广泛种植甘薯。尼日尔、尼日利亚和贝宁拥有丰富多样的甘薯种质资源,但其潜力迄今尚未得到充分探索。多样性阵列技术(DArTseq)是一种基于测序的基因分型(GBS)方法,已被开发出来,能够在不同作物物种中进行高密度单核苷酸多态性(SNP)基因分型。本研究的目的是通过基于测序的基因分型(GBS),利用多样性阵列技术评估西非甘薯种质资源的遗传多样性和群体结构。
使用29,523个多样性阵列技术(DArTseq)单核苷酸多态性标记对来自271份甘薯种质进行基因分型。遗传多样性分析显示,平均多态信息含量(PIC)值为0.39,次要等位基因频率为0.26,观察到的杂合度为10%。在第4号染色体上观察到最高的多态信息含量(PIC)值(0.41),而在第11号染色体上观察到最高比例的杂合度(He)(0.18)。分子多样性显示多态性位点(Ps)、θ(θ)和核苷酸多样性(π)的值较高,分别为0.973、0.158和0.086,这表明存在较高的遗传变异。成对遗传距离显示范围为0.08至0.47,总体平均值为0.34。群体结构分析在K = 4时将271份种质分为四个群体(群体1的特征是来自所有国家的种质混合;群体2主要由尼日利亚的育种系组成;群体3仅包含来自贝宁的地方品种;群体4仅由来自西非国家的地方品种组成),基于PhiPT值的分子方差分析(AMOVA)表明,当根据K = 4时的群体结构(25.25%)和聚类分析(19.43%)对种质进行分类时,大部分变异得到了解释。遗传距离显示,第4组(由尼日尔和贝宁的地方品种组成)与第2组(由75%的尼日利亚育种系组成)在遗传上距离较远(0.428),而第1组与第2组最接近(0.182)。
本研究使用了7,591个基于DArTseq的SNP标记,揭示了群体内部和群体之间广泛的多态性和变异。原产地国家之间的变异性(11.42%)超过了基于生物学状态(9.13%)和贮藏根肉色(7.90%)的变异性,强调了迁移对遗传多样性的影响。使用主成分分析(PCA)、邻接法(NJ)树和K = 4时的STRUCTURE进行的群体结构分析将271份种质分为不同的聚类,无论其地理起源如何,这表明存在广泛的遗传交换。第4组以地方品种为主(95%)与主要由育种系组成的第2组表现出显著的遗传分化(Nei's Gst = 0.428),表明它们作为杂交优势群在如HEBS或ABS等育种计划中的潜力。