Lu Yi, Lu Dongqing, Li Chujie, Chen Luping
Shanghai Tufeng Pharmaceutical Technology Co., Ltd., Shanghai 201203, China.
Jiangsu Kanion Pharmaceutical Co., Ltd., Lianyungang 222001, China.
Genes (Basel). 2024 Nov 29;15(12):1548. doi: 10.3390/genes15121548.
BACKGROUND/OBJECTIVES: Ulcerative colitis (UC) is a chronic inflammatory bowel disease (IBD) with a relapsing nature and complex etiology. Bioinformatics analysis has been widely applied to investigate various diseases. This study aimed to identify crucial differentially expressed genes (DEGs) and explore potential therapeutic agents for UC.
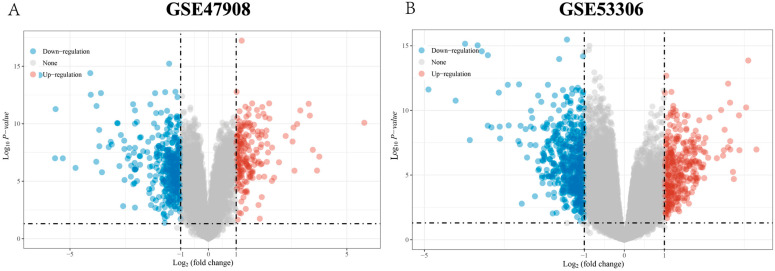
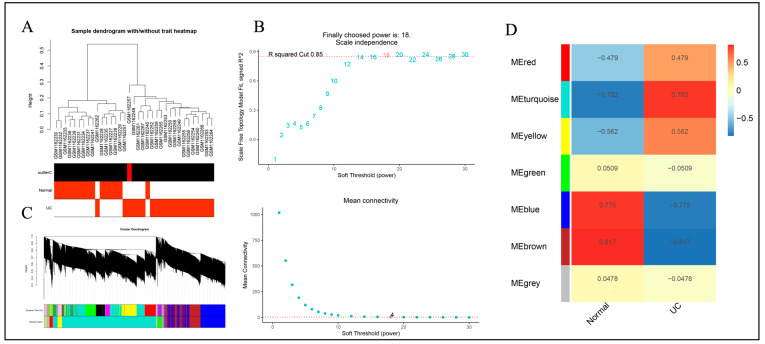
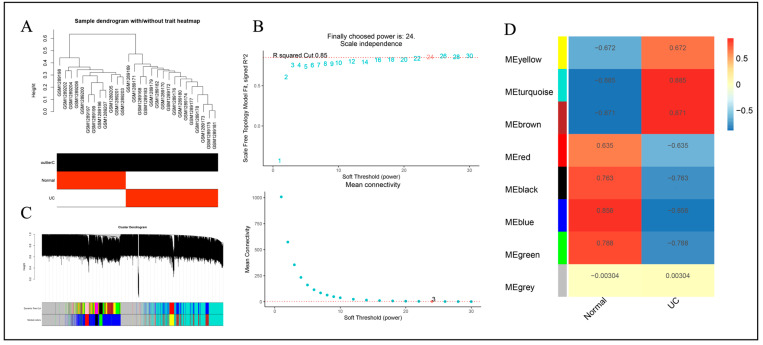
The GSE47908 and GSE55306 colon tissue transcriptome gene datasets were downloaded from the Gene Expression Omnibus-NCBI (GEO) database. GEO2R and Gene Set Enrichment Analysis (GSEA) were used to screen for DEGs in patients with UC compared to the normal population based on weighted gene co-expression network analysis (WGCNA). GO-BP analysis and KEGG enrichment analysis were performed on the intersecting differential genes via the Metascape website, while hub genes were analyzed by STRING11.0 and Cytoscape3.7.1. The expression of hub genes was verified in the dataset GSE38713 colon tissue specimens. Finally, the gene expression profiles of the validation set were analyzed by immuno-infiltration through the ImmuCellAI online tool, and the CMap database was used to screen for negatively correlated small molecule compounds.
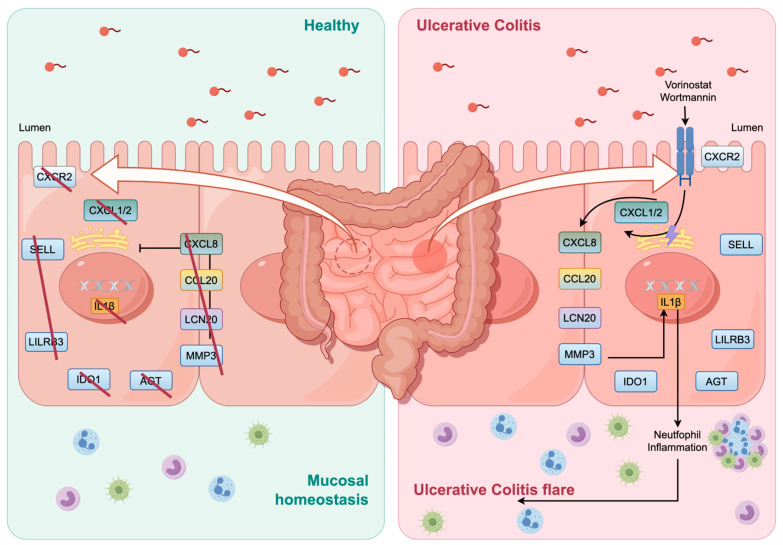
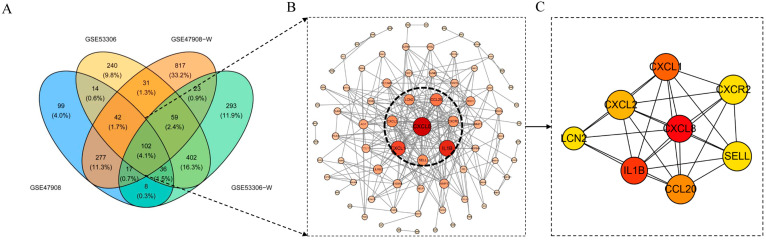
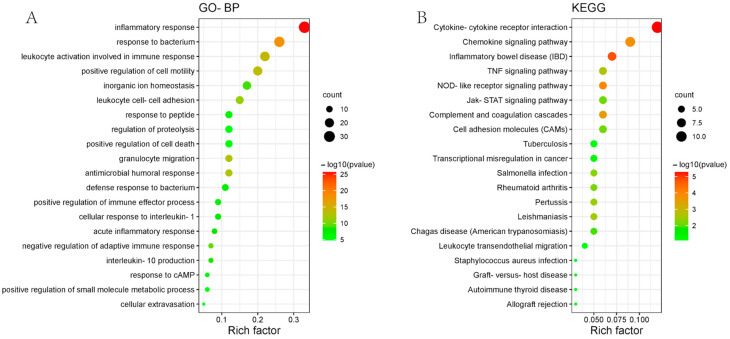
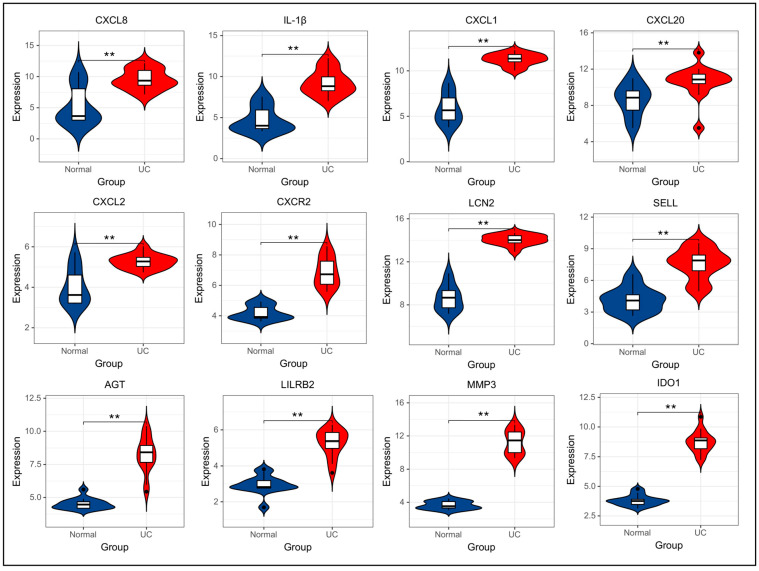
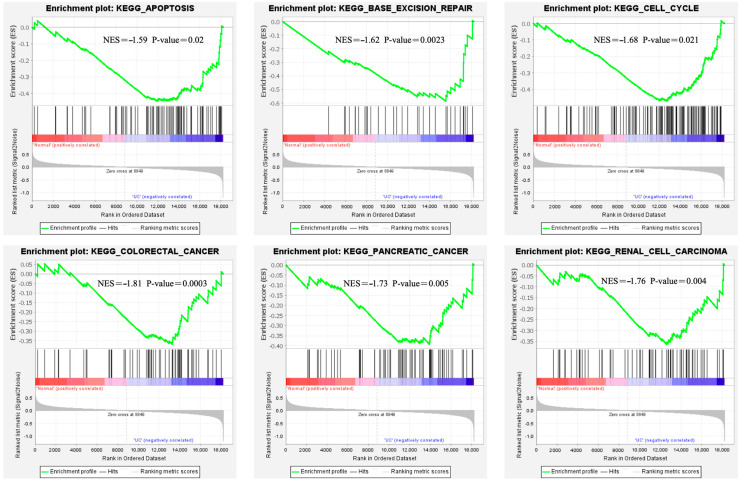
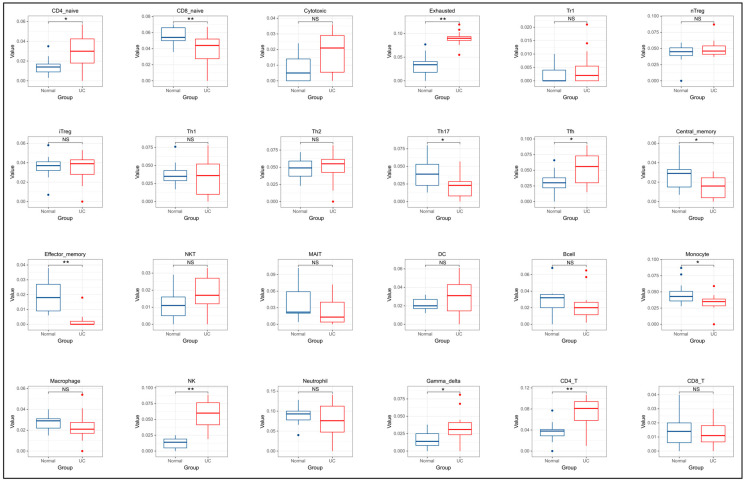
A total of 595 and 926 genes were screened by analysis of GSE47908 and GSE55306 datasets, respectively. Combined WGCNA hub module intersection yielded 12 hub genes (CXCL8, IL1β, CXCL1, CCL20, CXCL2, CXCR2, LCN2, SELL, AGT, LILRB3, MMP3, IDO1) associated with the pathogenesis of UC. GSEA analysis yielded intersecting pathways for both datasets (colorectal cancer pathway, base excision repair, cell cycle, apoptosis). GO-BP and KEGG enrichment analyses were performed to obtain key biological processes (inflammatory response, response to bacteria, leukocyte activation involved in the immune response, leukocyte-cell adhesion, apoptosis, positive regulation of immune effector processes) and key signaling pathways (cytokine-cytokine receptor interactions, IBD, NOD-like receptor signaling pathways). The immune cell infiltration analysis suggested that the incidence of UC was mainly related to the increase in CD4+T cells, depletion of T cells, T follicular helper cells, natural killer cells, γδ T cells and the decrease in CD8 naive T cells, helper T cells 17 and effector T cells. The CMap database results showed that small molecule compounds such as vorinostat, roxarsone, and wortmannin may be therapeutic candidates for UC.
This study not only aids in early prediction and prevention but also provides novel insights into the pathogenesis and treatment of UC.
背景/目的:溃疡性结肠炎(UC)是一种具有复发特性和复杂病因的慢性炎症性肠病(IBD)。生物信息学分析已广泛应用于各种疾病的研究。本研究旨在鉴定关键的差异表达基因(DEG)并探索UC的潜在治疗药物。
从美国国立生物技术信息中心(NCBI)的基因表达综合数据库(GEO)下载GSE47908和GSE55306结肠组织转录组基因数据集。基于加权基因共表达网络分析(WGCNA),使用GEO2R和基因集富集分析(GSEA)筛选UC患者与正常人群相比的DEG。通过Metascape网站对交集差异基因进行GO-BP分析和KEGG富集分析,同时通过STRING11.0和Cytoscape3.7.1分析枢纽基因。在数据集GSE38713结肠组织标本中验证枢纽基因的表达。最后,通过ImmuCellAI在线工具对验证集的基因表达谱进行免疫浸润分析,并使用CMap数据库筛选负相关的小分子化合物。
通过对GSE47908和GSE55306数据集的分析,分别筛选出595个和926个基因。联合WGCNA枢纽模块交集产生12个与UC发病机制相关的枢纽基因(CXCL8、IL1β、CXCL1、CCL20、CXCL2、CXCR2、LCN2、SELL、AGT、LILRB3、MMP3、IDO1)。GSEA分析得出两个数据集的交集通路(结直肠癌通路、碱基切除修复、细胞周期、凋亡)。进行GO-BP和KEGG富集分析以获得关键生物学过程(炎症反应、对细菌的反应、免疫反应中涉及的白细胞活化、白细胞-细胞粘附、凋亡、免疫效应过程的正调控)和关键信号通路(细胞因子-细胞因子受体相互作用、IBD、NOD样受体信号通路)。免疫细胞浸润分析表明,UC的发病主要与CD4 + T细胞增加、T细胞耗竭、滤泡辅助性T细胞、自然杀伤细胞、γδT细胞有关,以及CD8幼稚T细胞、辅助性T细胞17和效应性T细胞减少有关。CMap数据库结果显示,伏立诺他、洛克沙胂和渥曼青霉素等小分子化合物可能是UC的治疗候选药物。
本研究不仅有助于早期预测和预防,还为UC的发病机制和治疗提供了新的见解。