Failla Pinella, Saccuzzo Lucia, Galesi Ornella, Greco Donatella, Barresi Vincenza, Amata Silvestra, Romano Corrado, Fichera Marco
Oasi Research Institute-IRCCS, via Conte Ruggero 73, 94018 Troina, Italy.
Department of Biomedical and Biotechnological Sciences, Section of Clinical Biochemistry and Medical Genetics, University of Catania, via Santa Sofia, 95123 Catania, Italy.
Int J Mol Sci. 2024 Dec 16;25(24):13471. doi: 10.3390/ijms252413471.
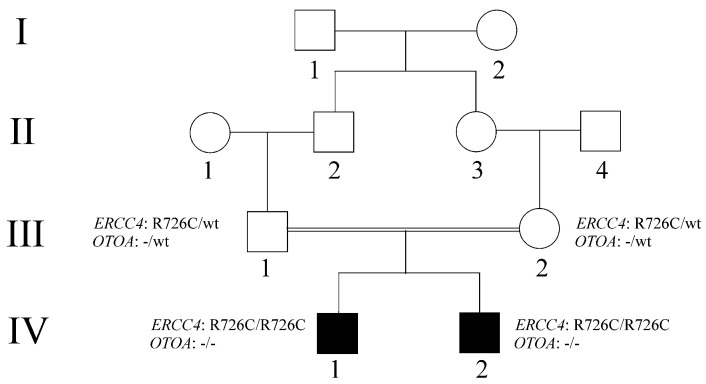
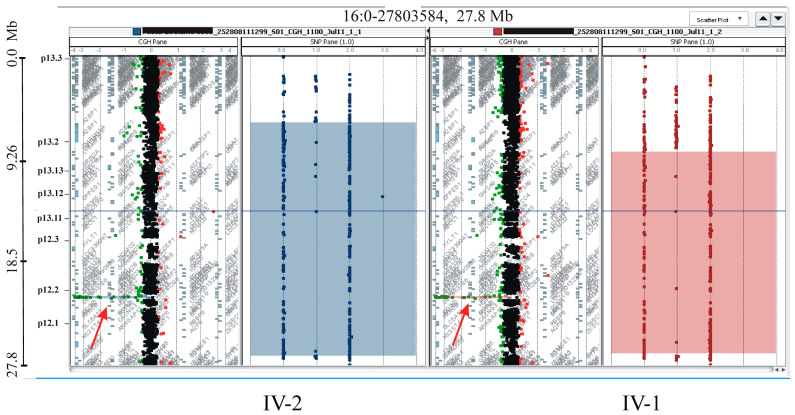
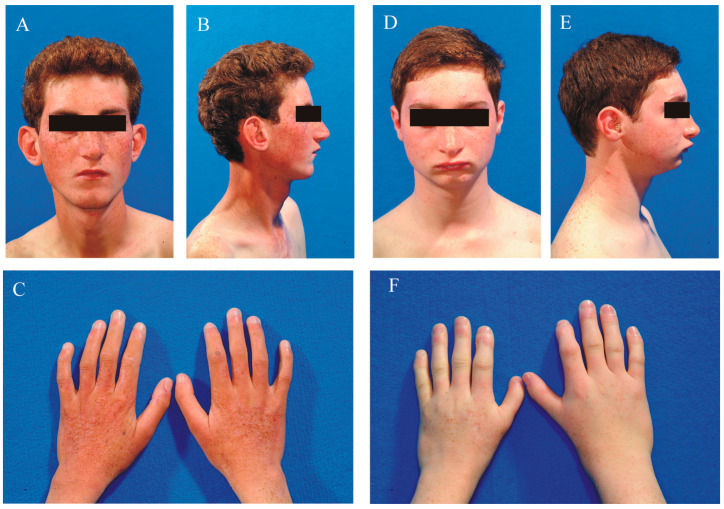
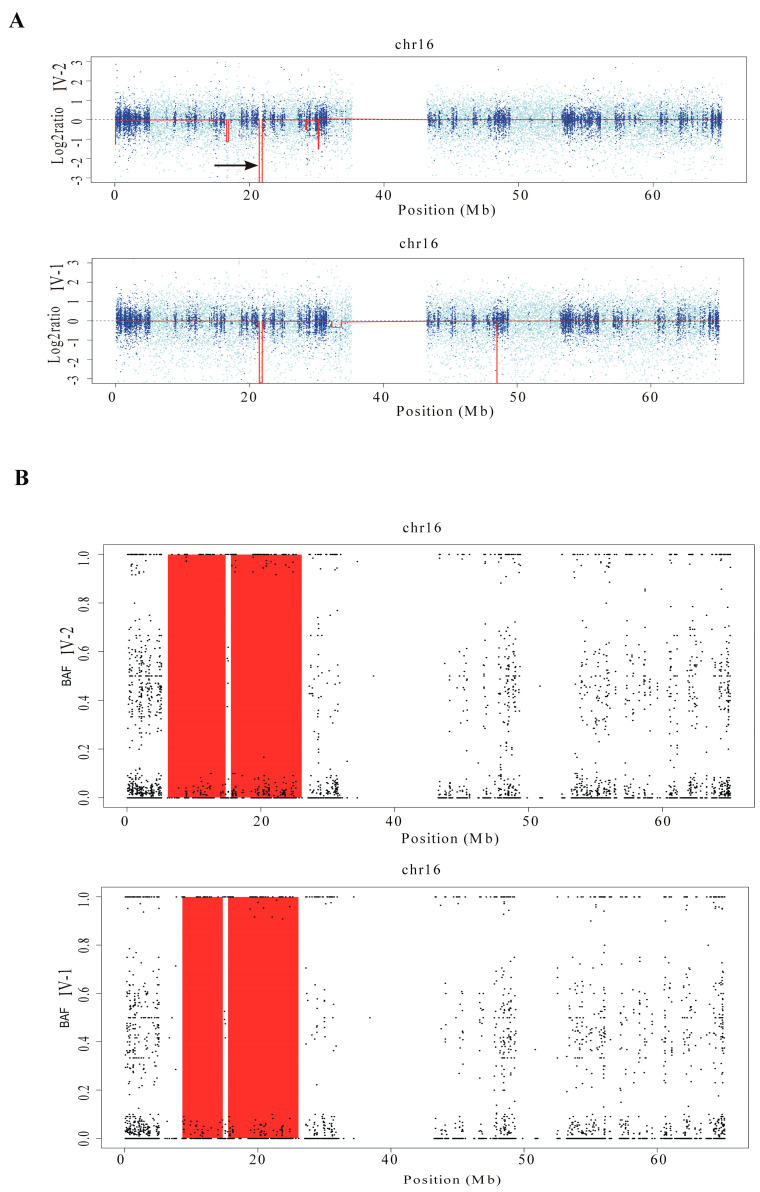
This study describes two siblings from consanguineous parents who exhibit intellectual disability, microcephaly, photosensitivity, bilateral sensorineural hearing loss, numerous freckles, and other clinical features that suggest a potential disruption of the nucleotide excision repair (NER) pathway. Whole exome sequencing (WES) identified a novel homozygous missense variant in the gene, which was predicted to be pathogenic. However, a subsequent peculiar audiometric finding prompted further investigation, revealing a homozygous deletion in the gene linked to neurosensorial hearing loss. Both variants were located within a run of homozygosity (ROH) on chromosome 16p13.12-p12.2, implicating a complex genetic basis for the observed phenotype. While this study reports a potentially novel variant, it underscores the importance of comprehensive analysis and deep phenotyping in WES data to improve diagnostic accuracy. Our findings advocate for an expanded approach in WES analysis, ensuring more precise diagnoses and improved genetic counseling, particularly when specialized tests for structural variant analysis are unavailable.
本研究描述了一对来自近亲父母的兄弟姐妹,他们表现出智力残疾、小头畸形、光敏性、双侧感音神经性听力损失、大量雀斑以及其他临床特征,提示核苷酸切除修复(NER)途径可能受到破坏。全外显子组测序(WES)在该基因中鉴定出一种新的纯合错义变异,预计具有致病性。然而,随后一项特殊的听力测定结果促使进一步调查,发现与神经感觉性听力损失相关的该基因存在纯合缺失。这两个变异均位于16号染色体p13.12 - p12.2的纯合子区域(ROH)内,提示所观察到的表型具有复杂的遗传基础。虽然本研究报告了一个潜在的新变异,但它强调了在WES数据中进行全面分析和深度表型分析以提高诊断准确性的重要性。我们的发现主张在WES分析中采用扩展方法,确保更精确的诊断和改善遗传咨询,特别是在没有用于结构变异分析的专门检测时。