Heidarpanah Sara, Li Kevin, Thibodeau Alexandre, Meniaï Ilhem, Parreira Valeria R, Quessy Sylvain, Segura Mariela, Fittipaldi Nahuel, Gaucher Marie-Lou
Chaire de Recherche en Salubrité des Viandes (CRSV), Département de Pathologie et Microbiologie, Faculté de Médecine Vétérinaire, Université de Montréal, Saint-Hyacinthe, QC J2S 2M2, Canada.
Swine and Poultry Infectious Diseases Research Centre (CRIPA), Faculté de Médecine Vétérinaire, Université de Montréal, Saint-Hyacinthe, QC J2S 2M2, Canada.
Microorganisms. 2024 Dec 18;12(12):2624. doi: 10.3390/microorganisms12122624.
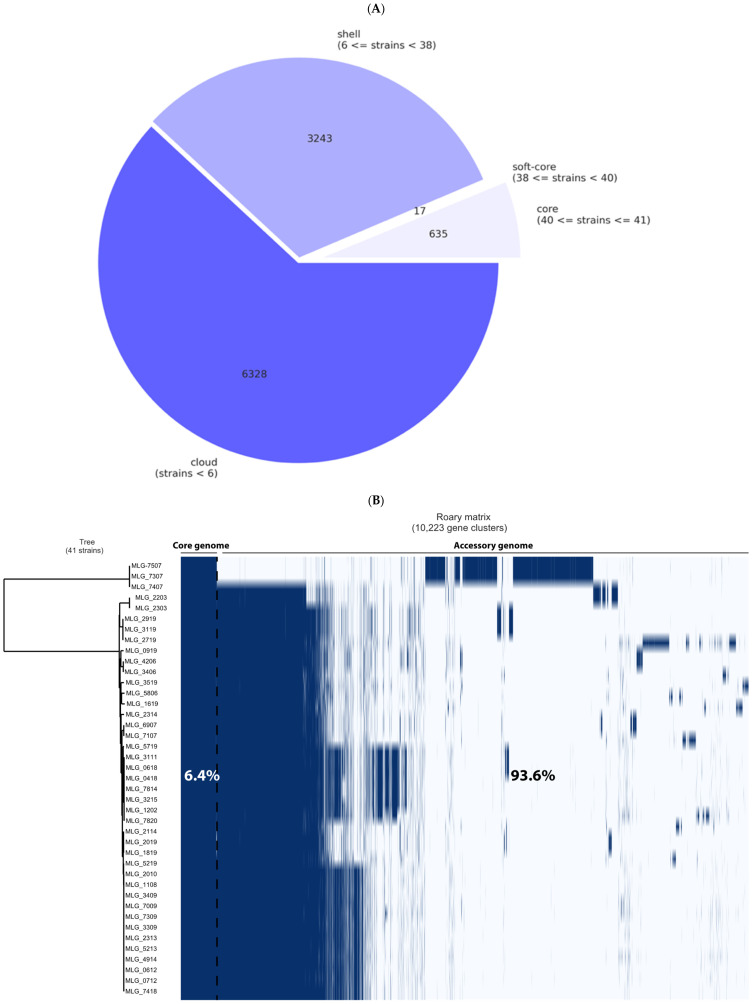
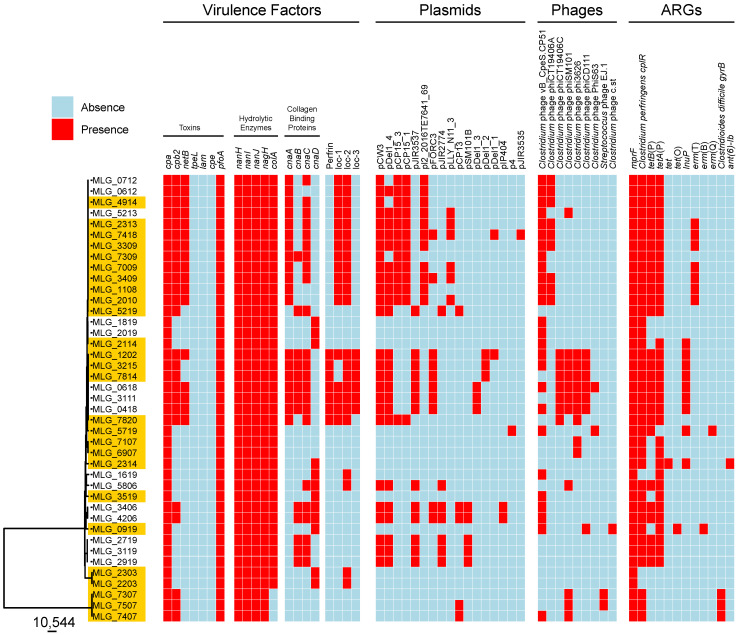
Avian necrotic enteritis due to the Gram-positive bacterium has re-emerged following the ban on antibiotic growth promoters in many poultry producing countries. The limited number of previous studies has left important gaps in our understanding of the genetic diversity and virulence traits of the pathogen. To address these knowledge gaps, in this study, we sequenced the genomes of 41 isolates recovered from commercial broiler chicken flocks in Quebec, Canada, including isolates from healthy birds and those affected by necrotic enteritis. We sought to understand the pangenome diversity and interrogated the genomes for key virulence factors involved in necrotic enteritis pathogenesis. On average, the genomes had a GC content of 28% and contained 3206 coding sequences. A variable presence of toxins, degradative hydrolytic enzymes, and collagen-binding proteins was also found. Through pangenome analysis, we revealed a total of 10,223 genes, 652 (6.4%) of which formed the core genome. Additionally, we identified 17 different plasmids, 12 antibiotic resistance genes, and nine prophage regions. Overall, our results demonstrated a relatively high genetic diversity among chicken isolates collected from the same geographical location, offering new insights into potential virulence mechanisms and adaptation of the pathogen within poultry populations.
由于革兰氏阳性菌导致的禽坏死性肠炎,在许多家禽生产国禁止使用抗生素生长促进剂之后再次出现。先前的研究数量有限,使得我们对该病原体的遗传多样性和毒力特性的理解存在重大空白。为了填补这些知识空白,在本研究中,我们对从加拿大魁北克的商业肉鸡群中分离出的41株菌株进行了基因组测序,其中包括从健康鸟类和受坏死性肠炎影响的鸟类中分离出的菌株。我们试图了解泛基因组多样性,并对参与坏死性肠炎发病机制的关键毒力因子的基因组进行研究。这些基因组的平均GC含量为28%,包含3206个编码序列。还发现了毒素、降解水解酶和胶原结合蛋白的可变存在情况。通过泛基因组分析,我们总共发现了10223个基因,其中652个(6.4%)构成了核心基因组。此外,我们鉴定出17种不同的质粒、12个抗生素抗性基因和9个前噬菌体区域。总体而言,我们的结果表明,从同一地理位置收集的鸡分离株之间存在相对较高的遗传多样性,为病原体在家禽群体中的潜在毒力机制和适应性提供了新的见解。