Yasuda Kasumi, Berenger Francois, Amaike Kazuma, Ueda Ayaka, Nakagomi Tomoya, Hamasaki Genki, Li Chen, Otani Noriko Yuyama, Kaitoh Kazuma, Tsuda Koji, Itami Kenichiro, Yamanishi Yoshihiro
Department of Bioscience and Bioinformatics, Kyushu Institute of Technology, 680-4 Kawazu, Iizuka, Fukuoka 820-8502, Japan.
Graduate School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwa-no-ha, Kashiwa, Chiba 277-8561, Japan.
iScience. 2024 Dec 17;28(1):111526. doi: 10.1016/j.isci.2024.111526. eCollection 2025 Jan 17.
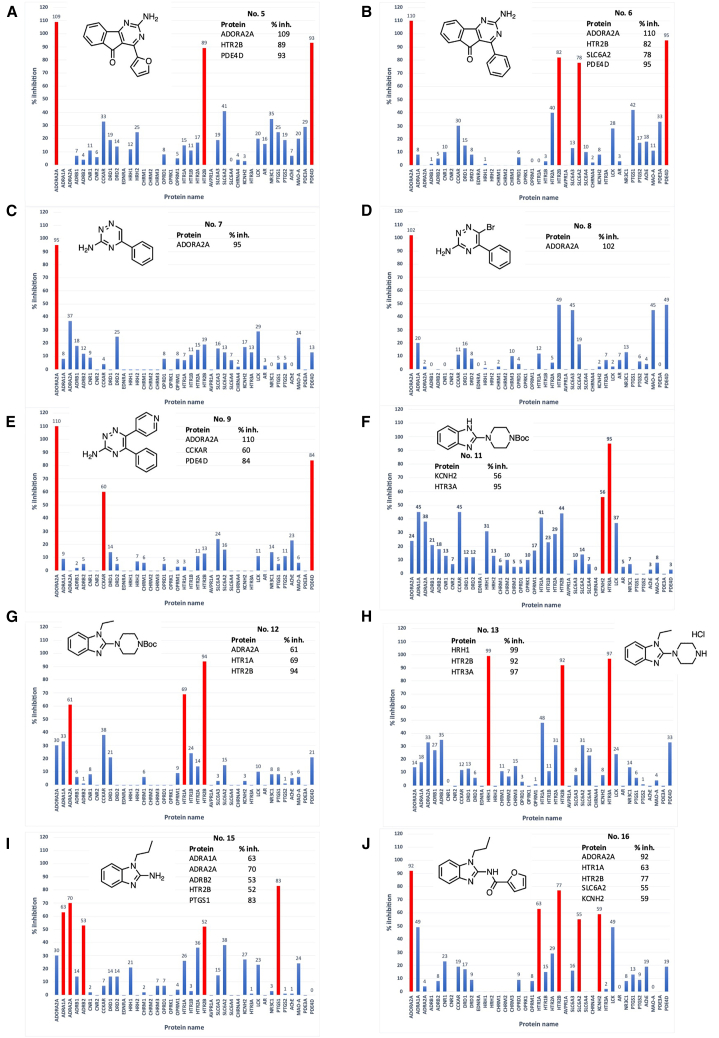
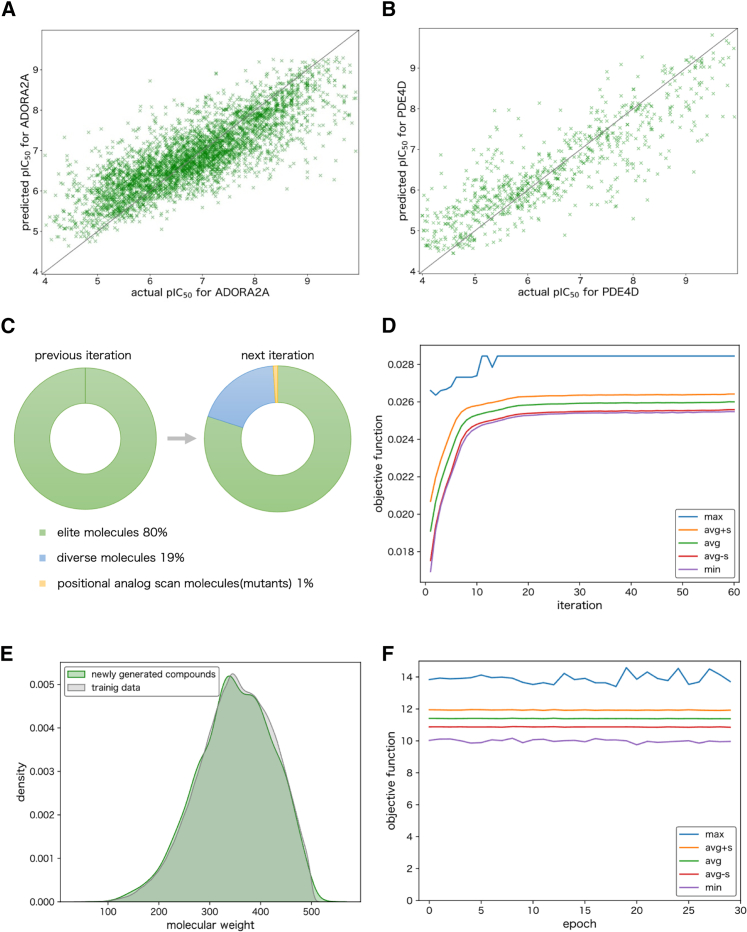
Drugs that interact with multiple therapeutic targets are potential high-value products in polypharmacology-based drug discovery, but the rational design remains a formidable challenge. Here, we present artificial intelligence (AI)-based methods to design the chemical structures of compounds that interact with multiple therapeutic target proteins. The molecular structure generation is performed by a fragment-based approach using a genetic algorithm with chemical substructures and a deep learning approach using reinforcement learning with stochastic policy gradients in the framework of generative adversarial networks. Using the proposed methods, we designed the chemical structures of compounds that would interact with two therapeutic targets of bronchial asthma, i.e., adenosine A2a receptor (ADORA2A) and phosphodiesterase 4D (PDE4D). We then synthesized 10 compounds and evaluated their bioactivities via the binding assays of 39 target human proteins, including ADORA2A and PDE4D. Three of the 10 synthesized compounds successfully interacted with ADORA2A and PDE4D with high specificity.
与多个治疗靶点相互作用的药物是基于多药理学的药物研发中潜在的高价值产品,但合理设计仍然是一项艰巨的挑战。在此,我们提出基于人工智能(AI)的方法来设计与多个治疗靶点蛋白相互作用的化合物的化学结构。分子结构生成通过基于片段的方法进行,该方法使用带有化学子结构的遗传算法,以及在生成对抗网络框架下使用带有随机策略梯度的强化学习的深度学习方法。使用所提出的方法,我们设计了与支气管哮喘的两个治疗靶点,即腺苷A2a受体(ADORA2A)和磷酸二酯酶4D(PDE4D)相互作用的化合物的化学结构。然后我们合成了10种化合物,并通过对包括ADORA2A和PDE4D在内的39种目标人类蛋白的结合试验评估了它们的生物活性。10种合成化合物中的3种成功地以高特异性与ADORA2A和PDE4D相互作用。