Liu Binbin, Xie Yumo, Zhang Yu, Tang Guannan, Lin Jinxin, Yuan Ze, Liu Xiaoxia, Wang Xiaolin, Huang Meijin, Luo Yanxin, Yu Huichuan
Department of Colorectal Surgery, The Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510655, Guangdong, China.
Guangdong Provincial Key Laboratory of Colorectal and Pelvic Floor Diseases, Guangdong Institute of Gastroenterology, The Sixth Affiliated Hospital, Sun Yat-sen University, 26 Yuancun Erheng Road, Guangzhou, 510655, Guangdong, China.
Cell Biosci. 2025 Jan 23;15(1):7. doi: 10.1186/s13578-024-01337-y.
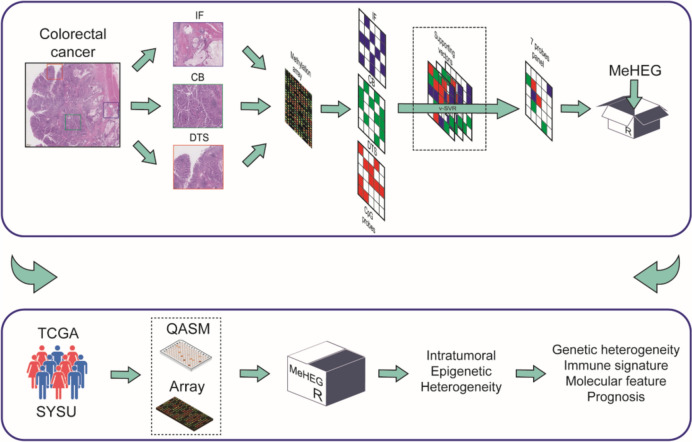
Intratumoral heterogeneity emerges from accumulating genetic and epigenetic changes during tumorigenesis, which may contribute to therapeutic failure and drug resistance. However, the lack of a quick and convenient approach to determine the intratumoral epigenetic heterogeneity (eITH) limit the application of eITH in clinical settings. Here, we aimed to develop a tool that can evaluate the eITH using the DNA methylation profiles from bulk tumors.
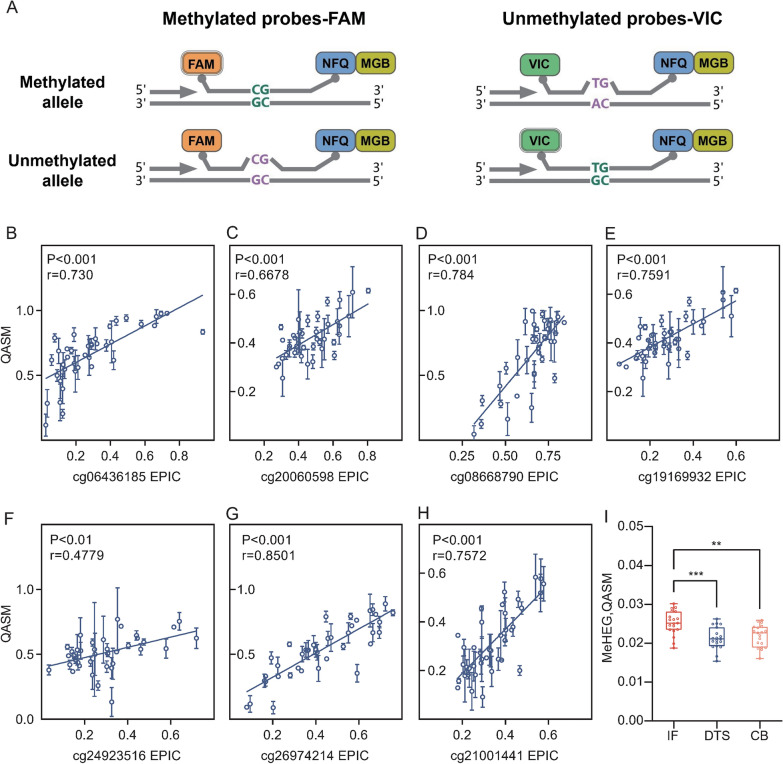
Genomic DNA of three laser micro-dissected tumor regions, including digestive tract surface, central bulk, and invasive front, was extracted from formalin-fixed paraffin-embedded sections of colorectal cancer patients. The genome-wide methylation profiles were generated with methylation array. The most variable methylated probes were selected to construct a DNA methylation-based heterogeneity (MeHEG) estimation tool that can deconvolve the proportion of each reference tumor region with the support vector machine model-based method. A PCR-based assay for quantitative analysis of DNA methylation (QASM) was developed to specifically determine the methylation status of each CpG in MeHEG assay at single-base resolution to realize fast evaluation of epigenetic heterogeneity.
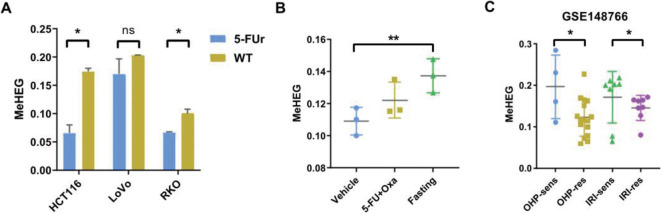
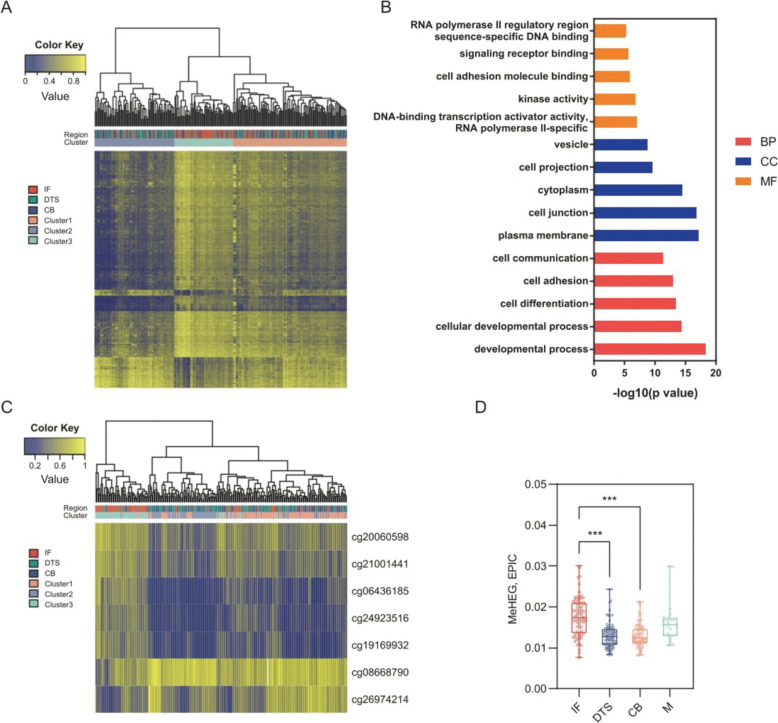
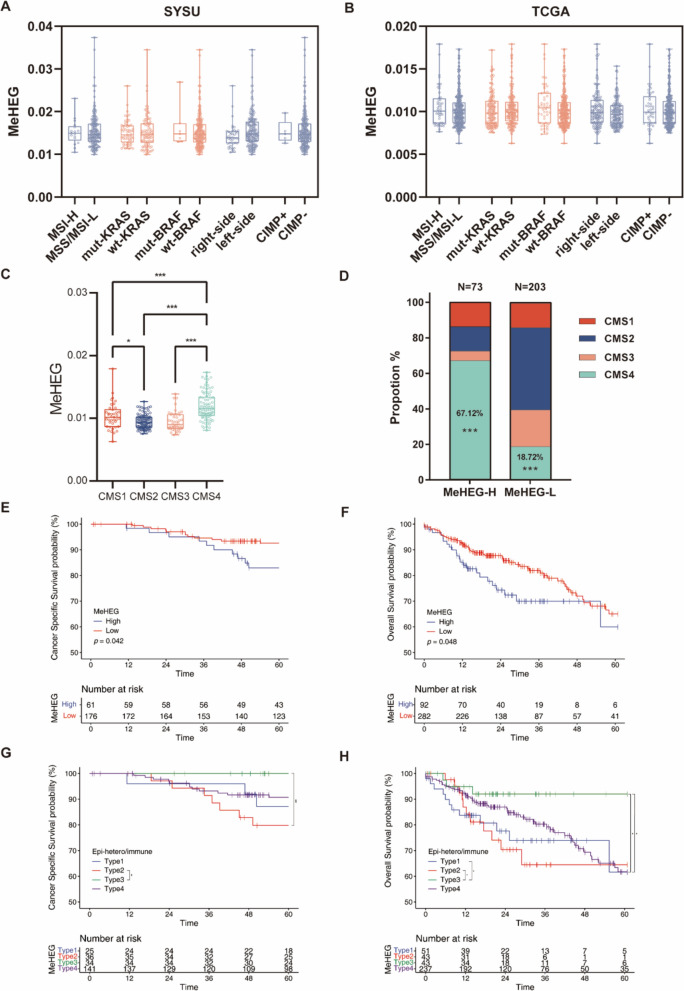
In the discovery set with 79 patients, the differentially methylated CpGs among the three tumor regions were found. The 7 most representative CpGs were identified and subsequently selected to develop the MeHEG algorithm. We validated its performance of deconvolution of tumor regions in an independent cohort. In addition, we showed the significant association of MeHEG-based epigenetic heterogeneity with the genomic heterogeneity in mutation and copy number variation in our in-house and TCGA cohorts. Besides, we found that the patients with higher MeHEG score had worse disease-free and overall survival outcomes. Finally, we found dynamic change of epigenetic heterogeneity based on MeHEG score in cancer cells under the treatment of therapeutic drugs.
By developing a 7-loci panel using a machine learning approach combined with the QASM assay for PCR-based application, we present a valuable method for evaluating intratumoral heterogeneity. The MeHEG algorithm offers novel insights into tumor heterogeneity from an epigenetic perspective, potentially enriching current knowledge of tumor complexity and providing a new tool for clinical and research applications in cancer biology.
肿瘤内异质性源于肿瘤发生过程中不断积累的基因和表观遗传变化,这可能导致治疗失败和耐药性。然而,缺乏一种快速便捷的方法来确定肿瘤内表观遗传异质性(eITH)限制了eITH在临床环境中的应用。在此,我们旨在开发一种工具,该工具可以使用来自大块肿瘤的DNA甲基化谱来评估eITH。
从结直肠癌患者的福尔马林固定石蜡包埋切片中提取三个激光显微切割肿瘤区域的基因组DNA,包括消化道表面、中央大块和浸润前沿。用甲基化芯片生成全基因组甲基化谱。选择变化最大的甲基化探针构建基于DNA甲基化的异质性(MeHEG)估计工具,该工具可以使用基于支持向量机模型的方法反卷积每个参考肿瘤区域的比例。开发了一种基于PCR的DNA甲基化定量分析方法(QASM),以单碱基分辨率特异性确定MeHEG分析中每个CpG的甲基化状态,以实现表观遗传异质性的快速评估。
在包含79例患者的发现集中,发现了三个肿瘤区域之间差异甲基化的CpG。鉴定出7个最具代表性的CpG,随后选择它们来开发MeHEG算法。我们在一个独立队列中验证了其对肿瘤区域反卷积的性能。此外,我们在我们的内部队列和TCGA队列中显示,基于MeHEG的表观遗传异质性与突变和拷贝数变异中的基因组异质性存在显著关联。此外,我们发现MeHEG评分较高的患者无病生存期和总生存期较差。最后,我们发现治疗药物处理下癌细胞中基于MeHEG评分的表观遗传异质性的动态变化。
通过使用机器学习方法结合基于PCR应用的QASM分析开发一个7位点面板,我们提出了一种评估肿瘤内异质性的有价值方法。MeHEG算法从表观遗传角度为肿瘤异质性提供了新的见解,可能丰富当前对肿瘤复杂性的认识,并为癌症生物学的临床和研究应用提供一种新工具。