Hu YaoFeng, Qin Sen, Deng RuCui
Department of Neurological Care Unit, The First Affiliated Hospital of YangTze University, Jingzhou, Hubei, China.
Department of Orthopedics, The First Affiliated Hospital of YangTze University, Jingzhou, Hubei, China.
Front Immunol. 2025 Jan 7;15:1512491. doi: 10.3389/fimmu.2024.1512491. eCollection 2024.
Recent years have seen persistently poor prognoses for glioma patients. Therefore, exploring the molecular subtyping of gliomas, identifying novel prognostic biomarkers, and understanding the characteristics of their immune microenvironments are crucial for improving treatment strategies and patient outcomes.
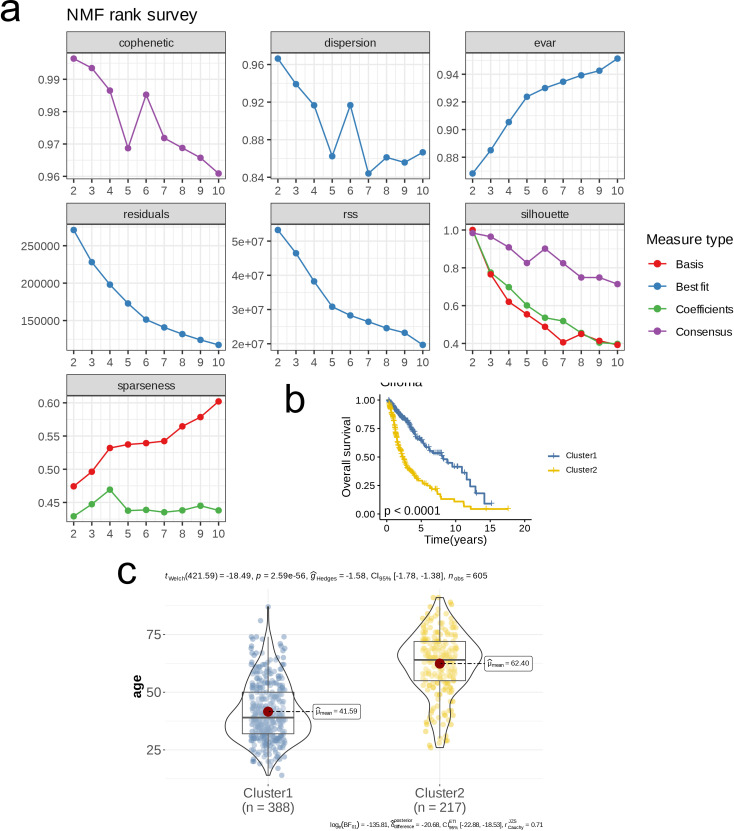
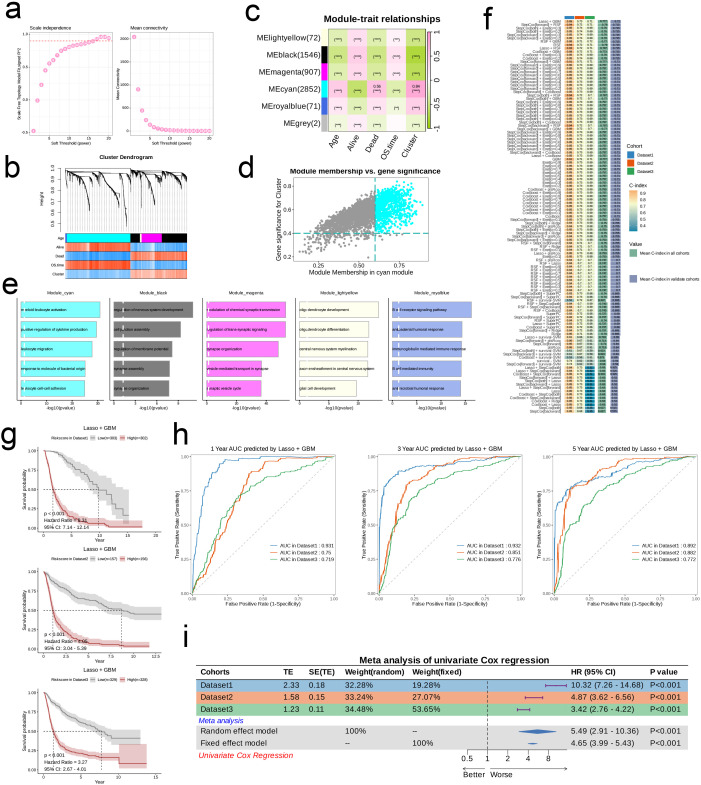
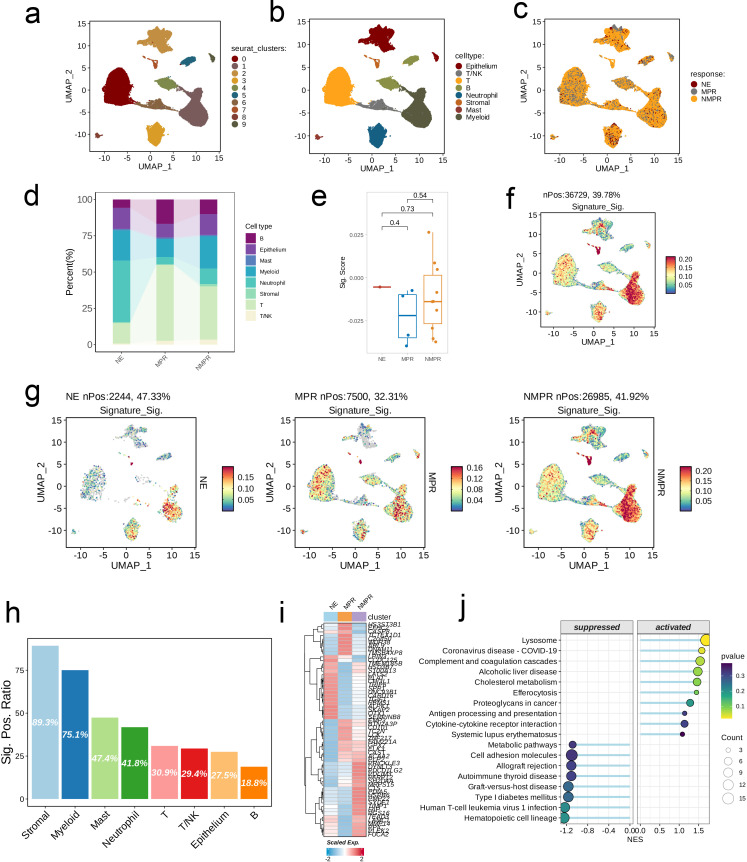
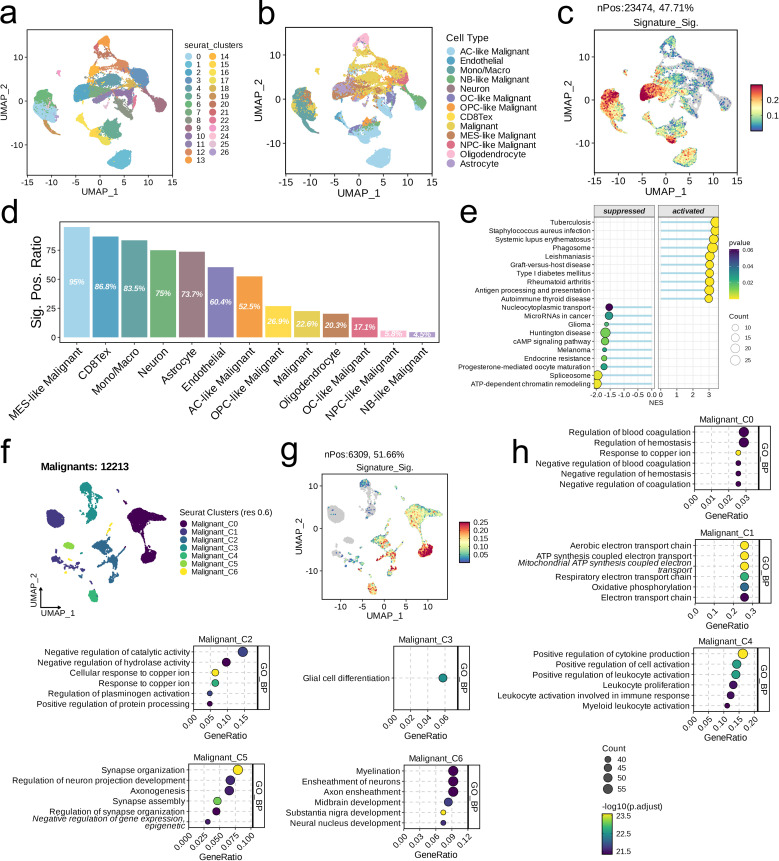
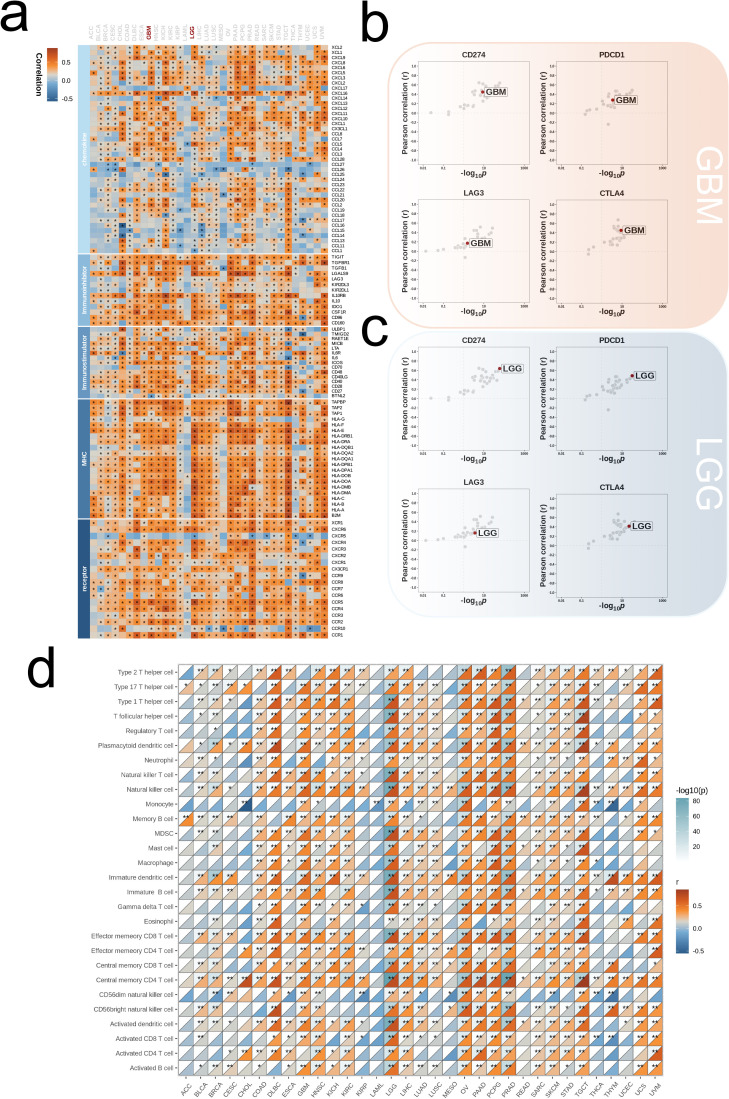
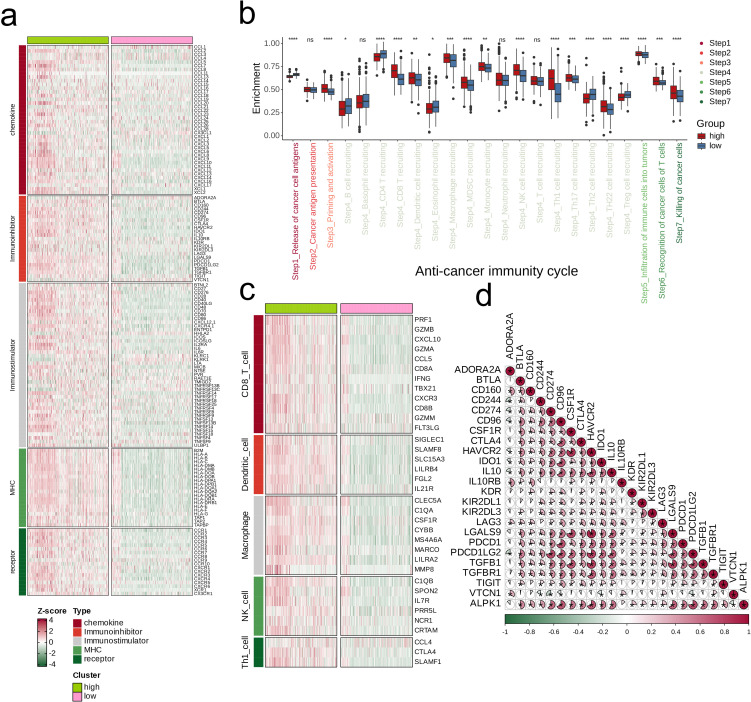
We integrated glioma datasets from multiple sources, employing Non-negative Matrix Factorization (NMF) to cluster samples and filter for differentially expressed metabolic genes. Additionally, we utilized Weighted Gene Co-expression Network Analysis (WGCNA) to identify key genes. A predictive model was developed utilizing the optimal consistency index derived from a combination of 101 machine learning techniques, and its effectiveness was confirmed through multiple datasets employing different methodologies. In-depth analyses were conducted on immune cell infiltration and tumor microenvironmental aspects. Single-cell sequencing data were employed for clustering and differential expression analysis of genes associated with glioma. Finally, the immune relevance of the model gene ALPK1 in the context of pan-cancer was explored, including its relationship with immune checkpoints.
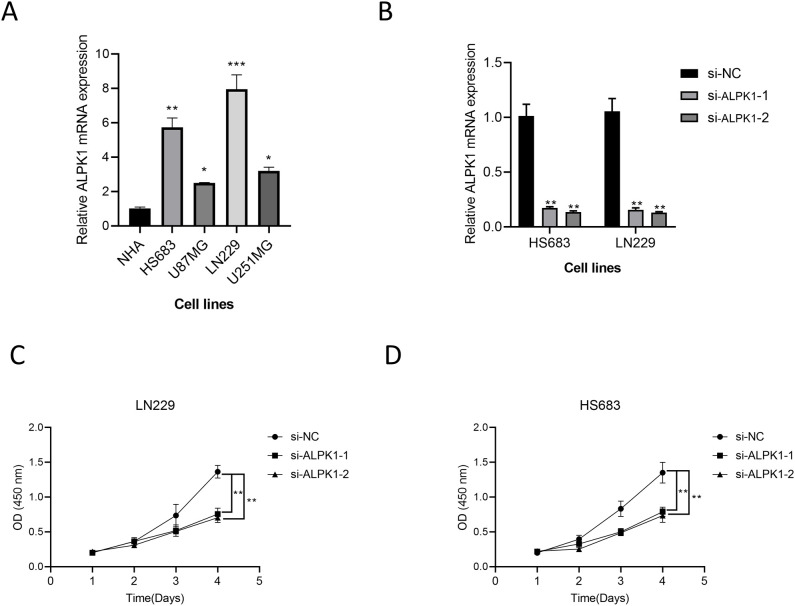
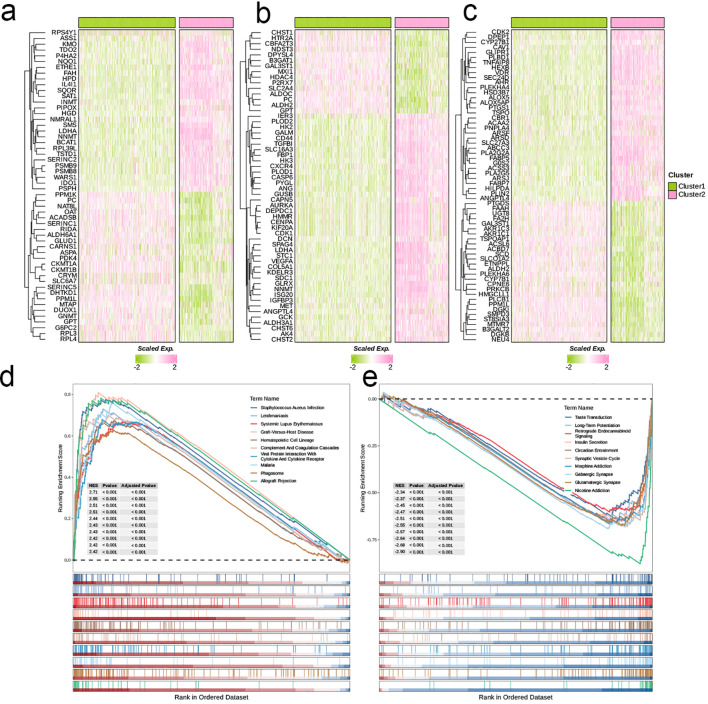
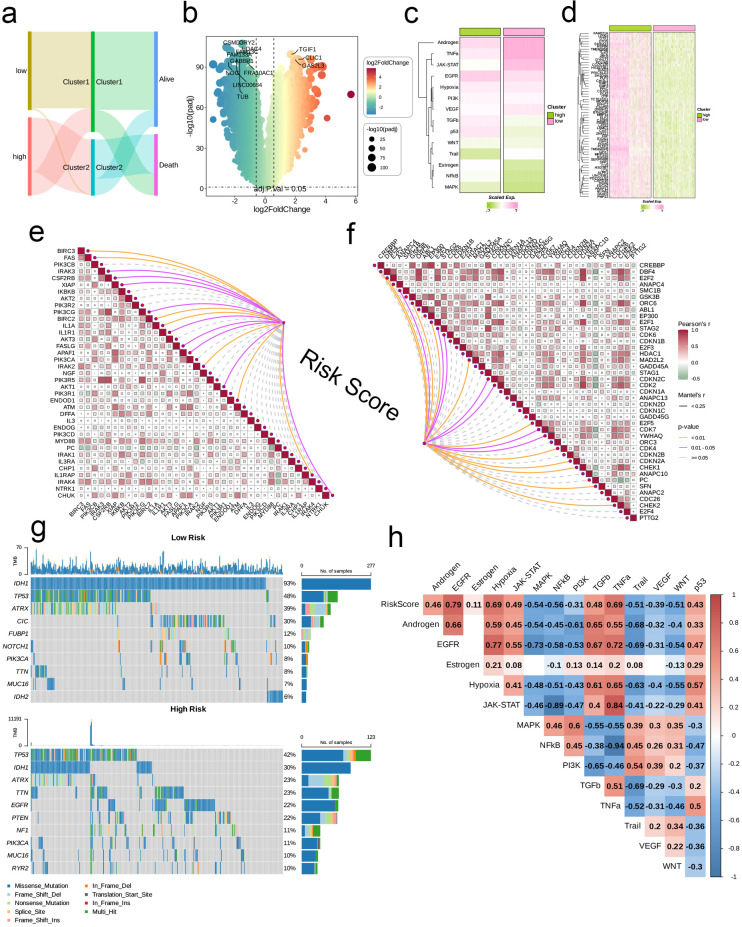
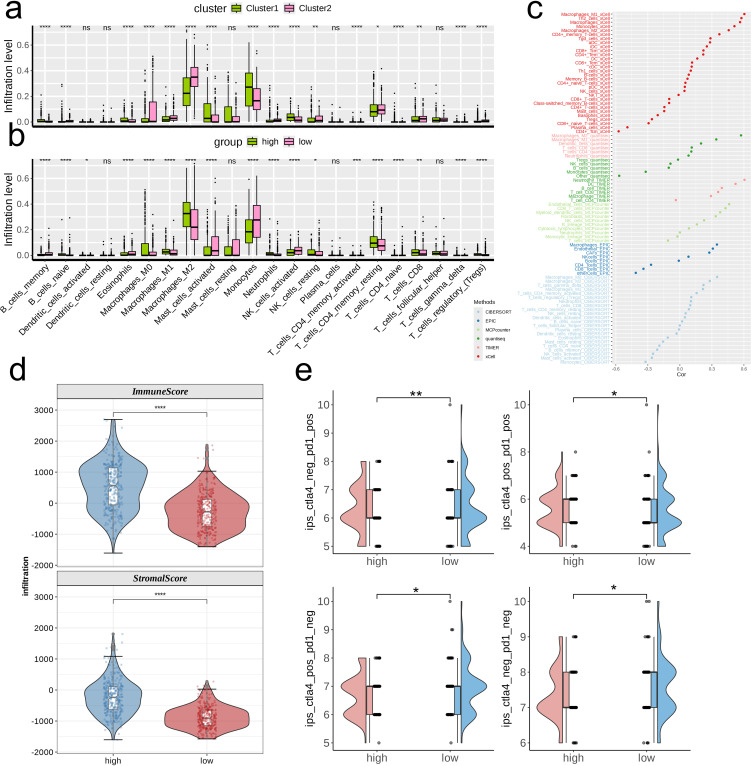
The application of NMF, coupled with differential analysis of metabolic-related genes, led to the identification of two clusters exhibiting significant differences in survival, age, and metabolic gene expression among patients. Core genes were identified through WGCNA, and a total of 101 machine learning models were constructed, with LASSO+GBM selected as the optimal model, demonstrating robust validation performance. Comprehensive analyses revealed that high-risk groups exhibited greater expression of specific genes, with ALPK1 showing significant correlations with immune regulation.
This research employed a multi-dataset strategy and various methods to clarify the differences in metabolic traits and immune conditions in glioma patients, while creating an innovative prognostic risk evaluation framework. These results offer fresh perspectives on the intricate biological processes that define gliomas.
近年来,胶质瘤患者的预后一直很差。因此,探索胶质瘤的分子亚型,识别新的预后生物标志物,并了解其免疫微环境的特征对于改善治疗策略和患者预后至关重要。
我们整合了来自多个来源的胶质瘤数据集,采用非负矩阵分解(NMF)对样本进行聚类,并筛选差异表达的代谢基因。此外,我们利用加权基因共表达网络分析(WGCNA)来识别关键基因。利用从101种机器学习技术组合中得出的最佳一致性指数开发了一个预测模型,并通过采用不同方法的多个数据集证实了其有效性。对免疫细胞浸润和肿瘤微环境方面进行了深入分析。利用单细胞测序数据对与胶质瘤相关的基因进行聚类和差异表达分析。最后,探讨了模型基因ALPK1在泛癌背景下的免疫相关性,包括其与免疫检查点的关系。
NMF的应用,结合代谢相关基因的差异分析,导致识别出两组在患者生存、年龄和代谢基因表达方面存在显著差异的集群。通过WGCNA确定了核心基因,并构建了总共101个机器学习模型,选择LASSO+GBM作为最佳模型,显示出强大的验证性能。综合分析表明,高危组特定基因的表达更高,ALPK1与免疫调节显著相关。
本研究采用多数据集策略和多种方法来阐明胶质瘤患者代谢特征和免疫状况的差异,同时创建了一个创新的预后风险评估框架。这些结果为定义胶质瘤的复杂生物学过程提供了新的视角。