Guccione Caitlin, Sfiligoi Igor, Gonzalez Antonio, Shaffer Justin P, Kazachkova Mariya, Weng Yuhan, McDonald Daniel, Shah Shailja C, Minot Samuel S, Paulson Thomas, Grady William M, Alexandrov Ludmil B, Knight Rob, Curtius Kit
Division of Biomedical Informatics, Department of Medicine, University of California San Diego, La Jolla, CA, USA.
Bioinformatics and Systems Biology Program, University of California San Diego, La Jolla, CA, USA.
bioRxiv. 2025 Jan 16:2025.01.14.633020. doi: 10.1101/2025.01.14.633020.
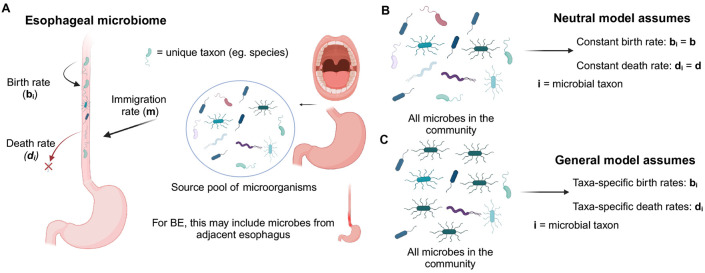
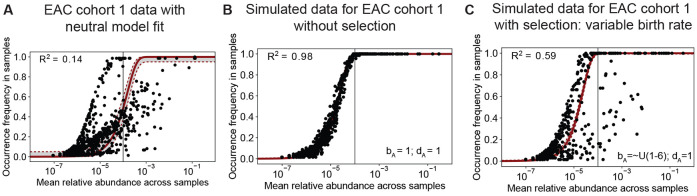
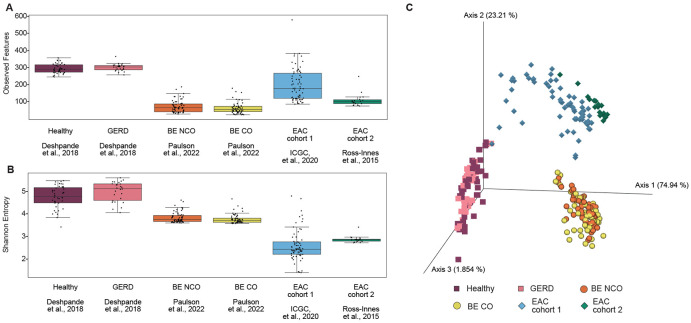
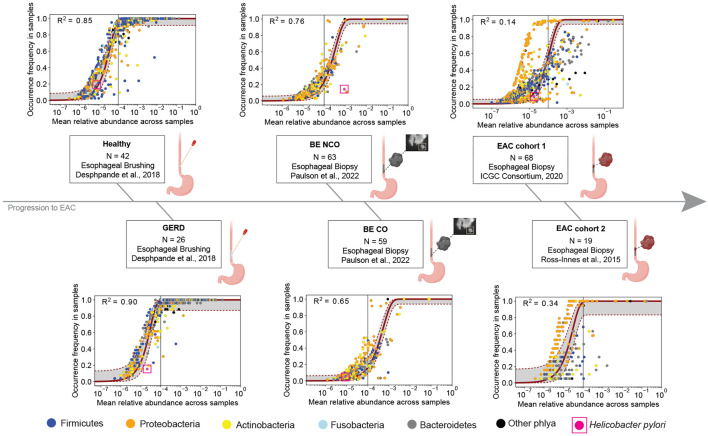
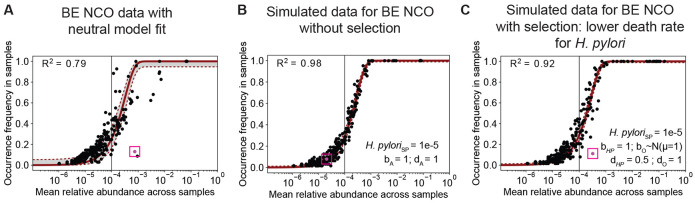
Mathematical modeling of somatic evolution, a process impacting both host cells and microbial communities in the human body, can capture important dynamics driving carcinogenesis. Here we considered models for esophageal adenocarcinoma (EAC), a cancer that has dramatically increased in incidence over the past few decades in Western populations, with high case fatality rates due to late-stage diagnoses. Despite advancements in genomic analyses of the precursor Barrett's esophagus (BE), prevention of late-stage EAC remains a significant clinical challenge. Previous microbiome studies in BE and EAC have focused on quantifying static microbial abundance differences rather than evolutionary dynamics. Using whole genome sequencing data from esophageal tissues, we first applied a robust bioinformatics pipeline to extract non-host DNA reads, mapped these putative reads to microbial taxa, and retained those taxa with high genomic coverage. When applying mathematical models of microbial evolution to sequential stages of progression to EAC, we observed evidence of neutral dynamics in community assembly within normal esophageal tissue and BE, but not EAC. In a case-control study of BE patients who progressed to EAC cancer outcomes (CO) versus those who had non-cancer outcomes (NCO) during follow-up (mean=10.5 years), we found that deviated significantly from the neutral expectation in BE NCO, suggesting that factors related to or infection itself may influence EAC risk. Additionally, simulations incorporating selection recapitulated non-neutral behaviors observed in the datasets. Formally modeling dynamics during progression holds promise in clinical applications by offering a deeper understanding of microbial involvement in cancer development.
体细胞进化的数学建模是一个影响人体宿主细胞和微生物群落的过程,它可以捕捉驱动致癌作用的重要动态。在这里,我们考虑了食管腺癌(EAC)的模型,在西方人群中,这种癌症的发病率在过去几十年中急剧上升,由于晚期诊断导致高病死率。尽管在癌前巴雷特食管(BE)的基因组分析方面取得了进展,但预防晚期EAC仍然是一项重大的临床挑战。先前关于BE和EAC的微生物组研究主要集中在量化静态微生物丰度差异,而非进化动态。利用食管组织的全基因组测序数据,我们首先应用了一个强大的生物信息学流程来提取非宿主DNA读数,将这些推定读数映射到微生物分类群,并保留那些具有高基因组覆盖率的分类群。当将微生物进化的数学模型应用于EAC进展的连续阶段时,我们在正常食管组织和BE中观察到群落组装中性动态的证据,但在EAC中没有。在一项对BE患者的病例对照研究中,在随访期间(平均 = 10.5年)进展为EAC癌症结局(CO)的患者与那些有非癌症结局(NCO)的患者相比,我们发现 在BE NCO中显著偏离中性预期,这表明与 或感染本身相关的因素可能影响EAC风险。此外,纳入选择的模拟重现了数据集中观察到的非中性行为。通过更深入地了解微生物在癌症发展中的作用,对进展过程中的动态进行形式化建模在临床应用中具有前景。