Sun Jinyuan, Zhu Tong, Cui Yinglu, Wu Bian
AIM Center, College of Life Sciences and Technology, Beijing University of Chemical Technology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China.
University of Chinese Academy of Sciences, Beijing, China.
Innovation (Camb). 2025 Jan 6;6(1):100750. doi: 10.1016/j.xinn.2024.100750.
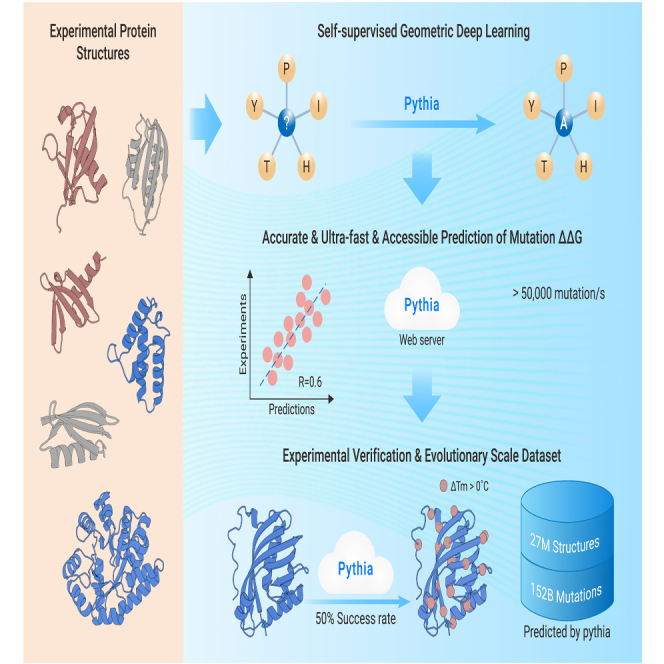
Predicting free energy changes (ΔΔG) is essential for enhancing our understanding of protein evolution and plays a pivotal role in protein engineering and pharmaceutical development. While traditional methods offer valuable insights, they are often constrained by computational speed and reliance on biased training datasets. These constraints become particularly evident when aiming for accurate ΔΔG predictions across a diverse array of protein sequences. Herein, we introduce Pythia, a self-supervised graph neural network specifically designed for zero-shot ΔΔG predictions. Our comparative benchmarks demonstrate that Pythia outperforms other self-supervised pretraining models and force field-based approaches while also exhibiting competitive performance with fully supervised models. Notably, Pythia shows strong correlations and achieves a remarkable increase in computational speed of up to 10-fold. We further validated Pythia's performance in predicting the thermostabilizing mutations of limonene epoxide hydrolase, leading to higher experimental success rates. This exceptional efficiency has enabled us to explore 26 million high-quality protein structures, marking a significant advancement in our ability to navigate the protein sequence space and enhance our understanding of the relationships between protein genotype and phenotype. In addition, we established a web server at https://pythia.wulab.xyz to allow users to easily perform such predictions.
预测自由能变化(ΔΔG)对于加深我们对蛋白质进化的理解至关重要,并且在蛋白质工程和药物开发中起着关键作用。虽然传统方法提供了有价值的见解,但它们常常受到计算速度和对有偏差训练数据集的依赖的限制。当旨在对各种蛋白质序列进行准确的ΔΔG预测时,这些限制变得尤为明显。在此,我们介绍了Pythia,这是一种专门为零样本ΔΔG预测设计的自监督图神经网络。我们的比较基准表明,Pythia优于其他自监督预训练模型和基于力场的方法,同时与全监督模型相比也表现出有竞争力的性能。值得注意的是,Pythia显示出很强的相关性,并且计算速度显著提高了高达10倍。我们进一步验证了Pythia在预测柠檬烯环氧水解酶的热稳定突变方面的性能,从而提高了实验成功率。这种卓越的效率使我们能够探索2600万个高质量的蛋白质结构,标志着我们在探索蛋白质序列空间以及加深对蛋白质基因型和表型之间关系的理解方面取得了重大进展。此外,我们在https://pythia.wulab.xyz建立了一个网络服务器,以便用户能够轻松地进行此类预测。