Xie Wuming, Liao Baoqiong, Shuai Mei, Liu Rutian, Hong Min, He Shuwen
Ganzhou People's Hospital, Ganzhou, Jiangxi, China.
Ganzhou Maternal and Child Health Hospital, Ganzhou, Jiangxi, China.
Mol Genet Genomic Med. 2025 Feb;13(2):e70066. doi: 10.1002/mgg3.70066.
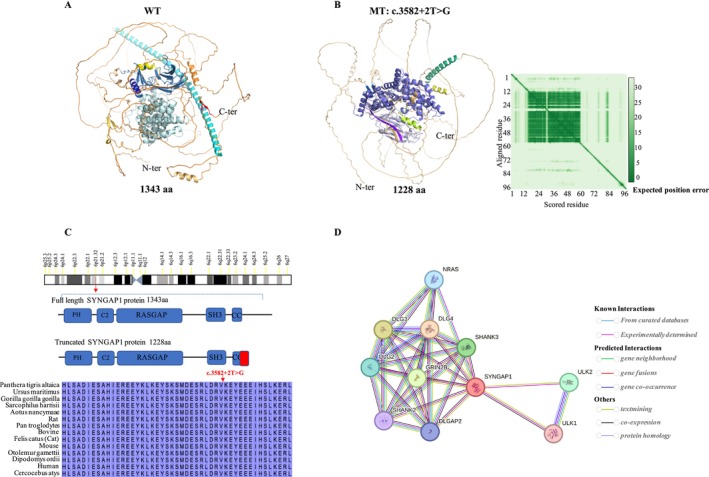
SYNGAP1 encodes a Ras/Rap GTPase-activating protein that is predominantly expressed in the brain with the functional roles in regulating synaptic plasticity, spine morphogenesis, and cognition function. Pathogenic variants in SYNGAP1 have been associated with a spectrum of neurodevelopmental disorders characterized by developmental delays, intellectual disabilities, epilepsy, hypotonia, and the features of autism spectrum disorder. The aim of this study was to identify a novel SYNGAP1 gene variant linked to neurodevelopmental disorders and to evaluate the pathogenicity of the detected variant.
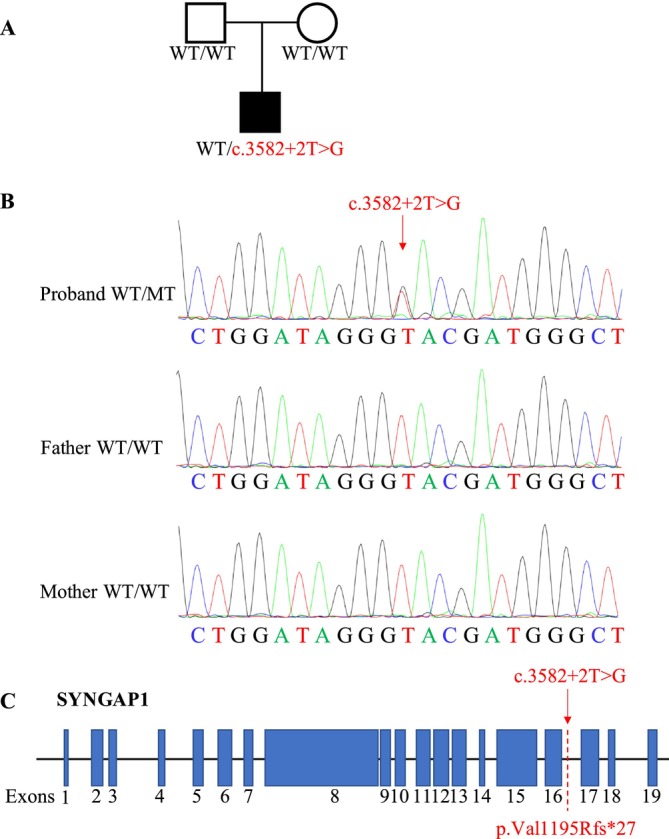
A novel de novo intronic variant in SYNGAP1 was identified by Whole exome sequencing (WES) and confirmed by Sanger sequencing. Minigene assays were conducted to assess whether the intronic variant in SYNGAP1 influenced the normal splicing of mRNA.
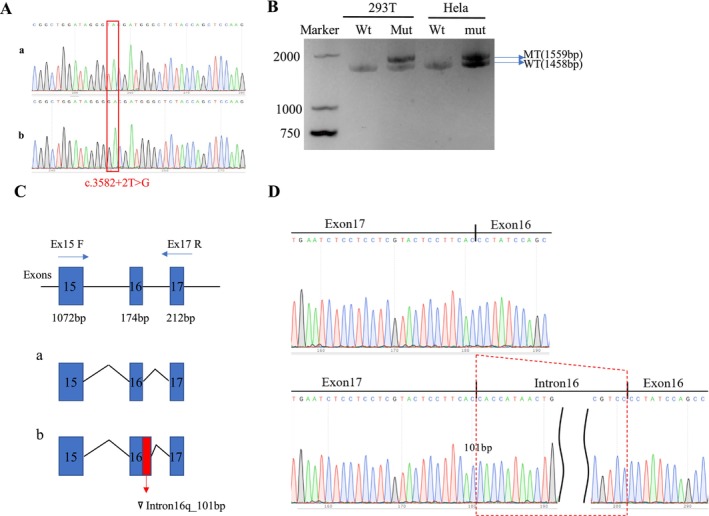
A novel de novo intronic variant in SYNGAP1 (c.3582+2T>G) was indentified with clinical features suggestive of neurodevelopmental related disorders. Minigene splicing analysis demonstrated that this noncanonical splice site variant led to the activation of a cryptic acceptor splice site. Consequently, 101 base pairs of intron 16 were aberrantly retained in the mRNA, leading to a frameshift. This frameshift resulted in the introduction of a premature stop codon (TGA) in the coding sequence and the production of a truncated SYNGAP1 protein, potentially leding to loss of function and subsequent disruption of its biological roles.
Our findings highlight the significance of de novo pathogenic SYNGAP1 variants at the intron 16/exon 17 junction in the SYNGAP1-related neurodevelopmental disorders, providing novel insights into the genetic basis and diagnosis of these disabilities.
SYNGAP1编码一种Ras/Rap GTP酶激活蛋白,主要在大脑中表达,在调节突触可塑性、树突棘形态发生和认知功能方面发挥作用。SYNGAP1中的致病变异与一系列神经发育障碍相关,这些障碍的特征包括发育迟缓、智力残疾、癫痫、肌张力减退以及自闭症谱系障碍的特征。本研究的目的是鉴定一种与神经发育障碍相关的新型SYNGAP1基因变异,并评估检测到的变异的致病性。
通过全外显子组测序(WES)鉴定出SYNGAP1中的一种新型从头内含子变异,并通过桑格测序进行确认。进行了小基因分析,以评估SYNGAP1中的内含子变异是否影响mRNA的正常剪接。
鉴定出SYNGAP1中的一种新型从头内含子变异(c.3582+2T>G),其临床特征提示与神经发育相关障碍。小基因剪接分析表明,这种非典型剪接位点变异导致了一个隐匿性受体剪接位点的激活。因此,第16号内含子的101个碱基对异常保留在mRNA中,导致移码。这种移码导致在编码序列中引入一个提前终止密码子(TGA),并产生截短的SYNGAP1蛋白,可能导致功能丧失并随后破坏其生物学作用。
我们的研究结果突出了SYNGAP1相关神经发育障碍中第16号内含子/第17号外显子交界处的从头致病性SYNGAP1变异的重要性,为这些残疾的遗传基础和诊断提供了新的见解。