Joseph Donald J, Mercado-Ayon Elizabeth, Flatley Liam, Viaene Angela N, Hordeaux Juliette, Marsh Eric D, Lynch David R
Division of Neurology, Department of Pediatrics, The Children's Hospital of Philadelphia, Philadelphia, PA, 19104, USA.
Department of Neurology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, 19104, USA.
Cerebellum. 2025 Feb 5;24(2):42. doi: 10.1007/s12311-025-01796-0.
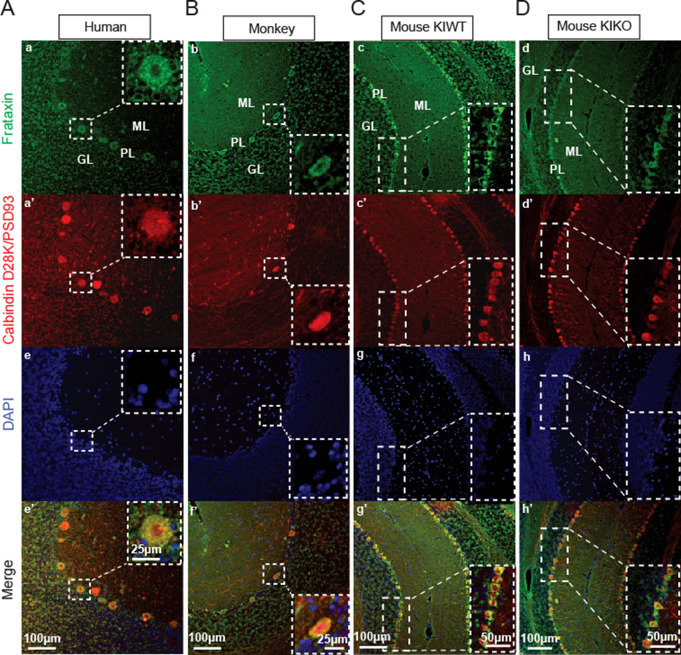
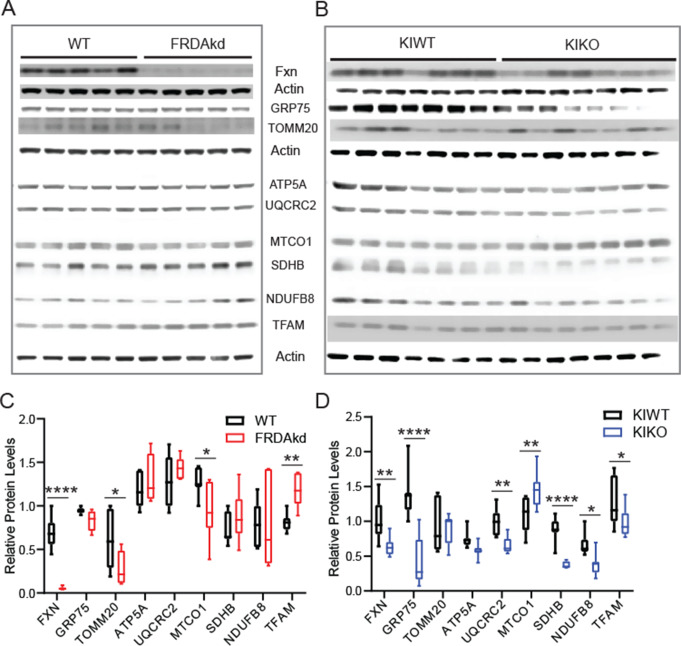
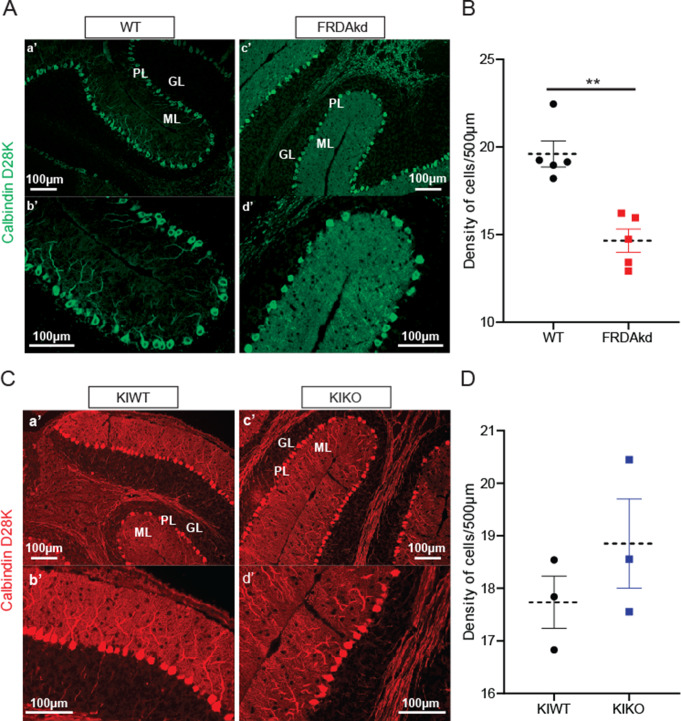
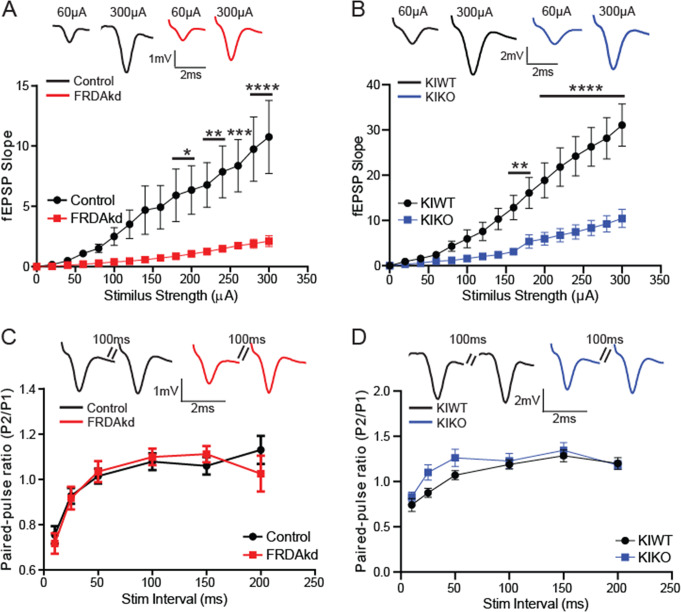
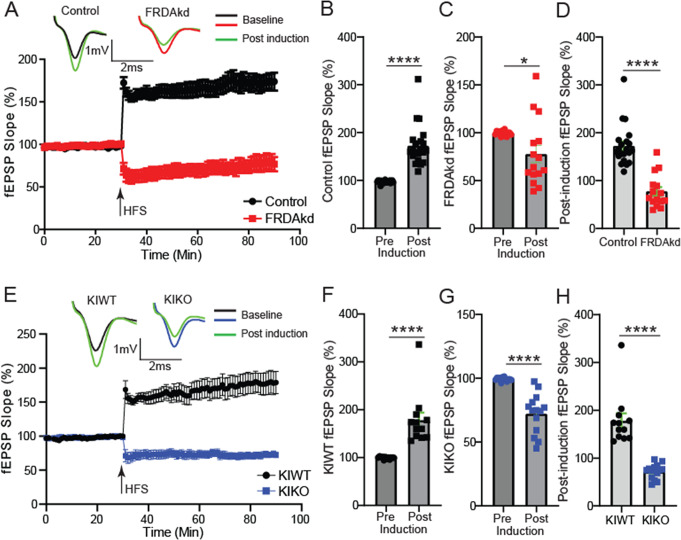
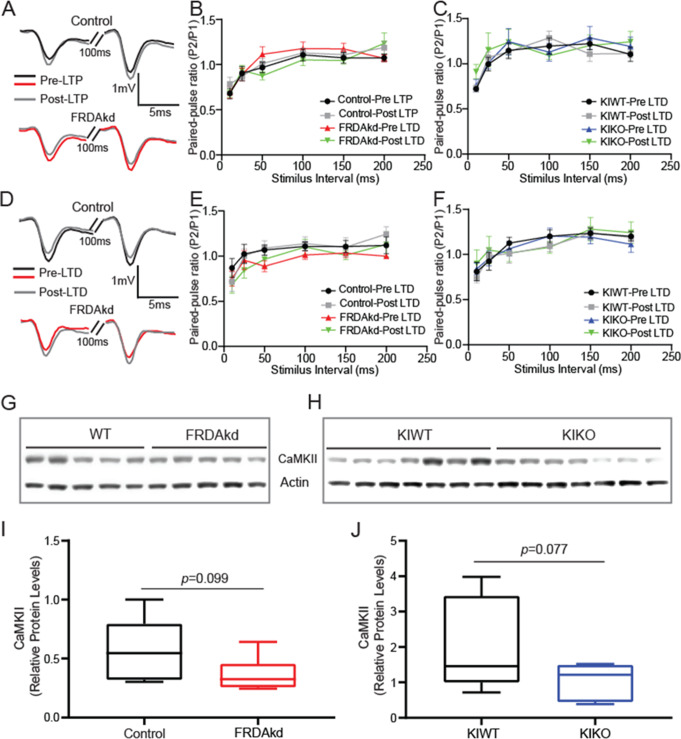
Friedreich ataxia (FRDA) is an autosomal recessive disorder caused by GAA expansions in the FXN gene, which codes for the protein frataxin (FXN). These mutations reduce FXN expression, leading to mitochondrial dysfunction and multisystemic disease. Accumulating evidence suggests that neuronal dysfunction, rather than neuronal death, may drive the neurological phenotypes of FRDA, but the mechanisms underlying such neurological phenotypes remain unclear. To investigate the neural circuit basis of this dysfunction, we employed field recordings to measure Purkinje cell (PC) function and synaptic properties along with western blotting and immunohistochemistry to determine their density and structure in two established FRDA mouse models, the shRNA-frataxin (FRDAkd) and the frataxin knock in-knockout (KIKO) mice. Western blotting demonstrated subtle changes in mitochondrial proteins and only a modest reduction in the density of calbindin positive cells PCs in the cerebellar cortex of the FRDAkd mice, with no change in the density of PCs in the KIKO mice. Though PC density differed slightly in the two models, field recordings of parallel fiber-PC synapses in the molecular layer demonstrated concordant hypo-excitability of basal synaptic transmission and impairments of long-term plasticity using induction protocols associated with both potentiation and depression of synaptic strength. These results indicate that synaptic instability might be a common feature in FRDA mouse models.
弗里德赖希共济失调(FRDA)是一种常染色体隐性疾病,由FXN基因中的GAA重复扩增引起,该基因编码铁调素(FXN)蛋白。这些突变会降低FXN的表达,导致线粒体功能障碍和多系统疾病。越来越多的证据表明,神经元功能障碍而非神经元死亡可能是FRDA神经表型的驱动因素,但这些神经表型背后的机制仍不清楚。为了研究这种功能障碍的神经回路基础,我们采用场电位记录来测量浦肯野细胞(PC)的功能和突触特性,并结合蛋白质免疫印迹法和免疫组织化学法来确定两种已建立的FRDA小鼠模型,即shRNA-铁调素(FRDAkd)小鼠和铁调素敲入敲除(KIKO)小鼠中PC的密度和结构。蛋白质免疫印迹法显示线粒体蛋白有细微变化,在FRDAkd小鼠的小脑皮质中,钙结合蛋白阳性PC的密度仅略有降低,而在KIKO小鼠中PC的密度没有变化。尽管在这两种模型中PC密度略有不同,但分子层中平行纤维-PC突触的场电位记录显示,基础突触传递存在一致的低兴奋性,并且使用与突触强度增强和减弱相关的诱导方案时,长期可塑性受损。这些结果表明,突触不稳定可能是FRDA小鼠模型的一个共同特征。