Mercado-Ayón Elizabeth, Warren Nathan, Halawani Sarah, Rodden Layne N, Ngaba Lucie, Dong Yi Na, Chang Joshua C, Fonck Carlos, Mavilio Fulvio, Lynch David R, Lin Hong
Department of Neurology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States.
Department of Pediatrics and Neurology, The Children's Hospital of Philadelphia, Philadelphia, PA, United States.
Front Neurosci. 2022 Mar 24;16:819569. doi: 10.3389/fnins.2022.819569. eCollection 2022.
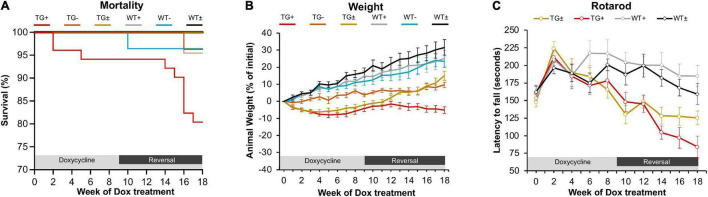
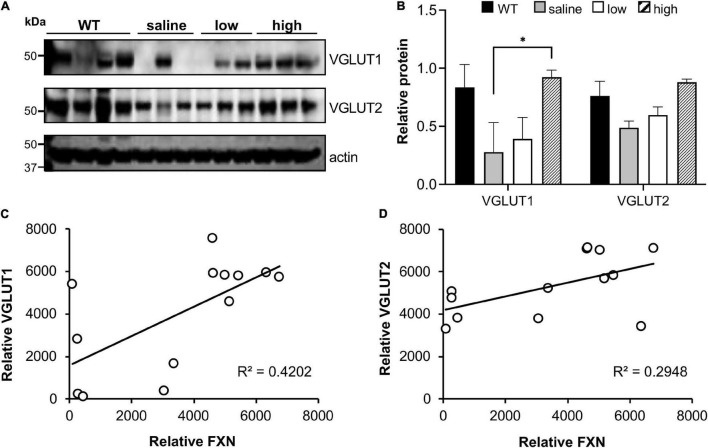
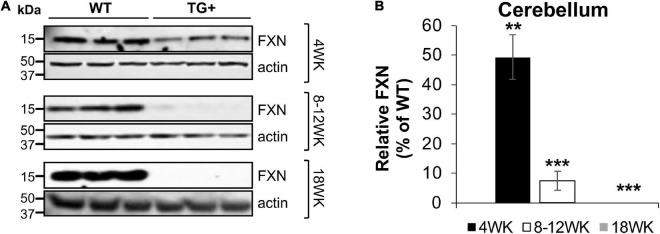
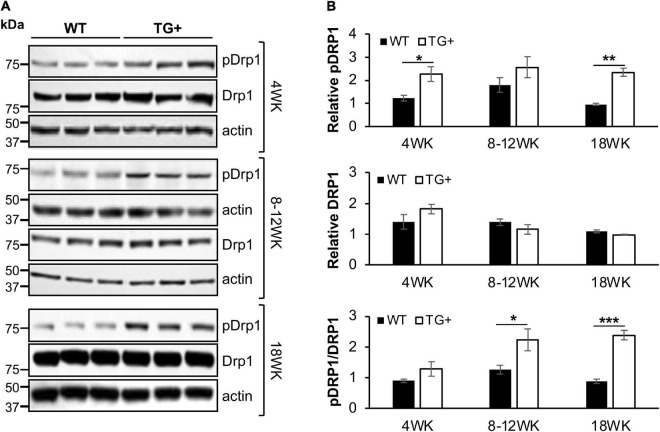
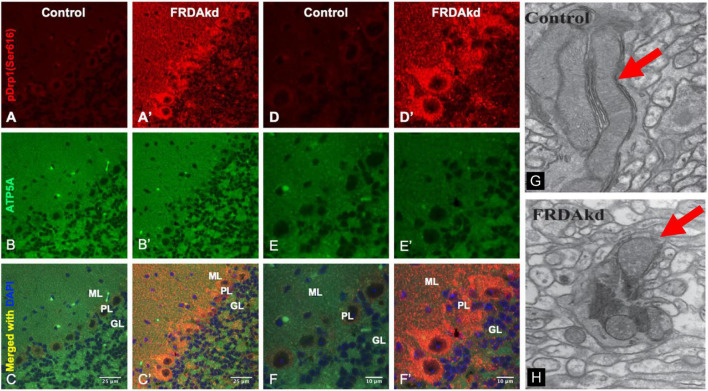
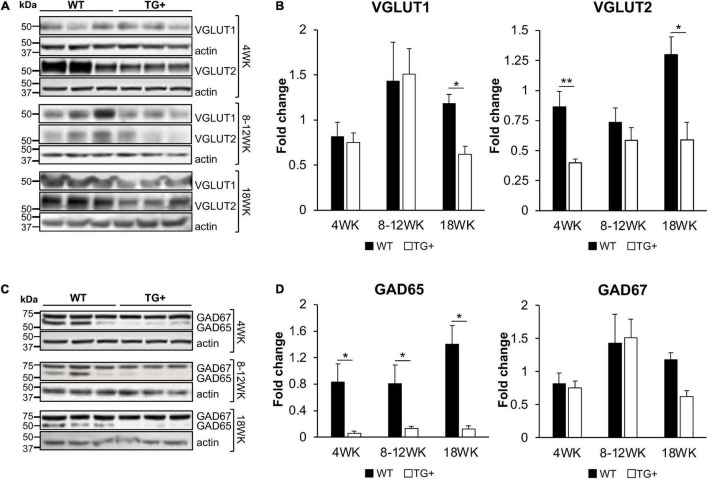
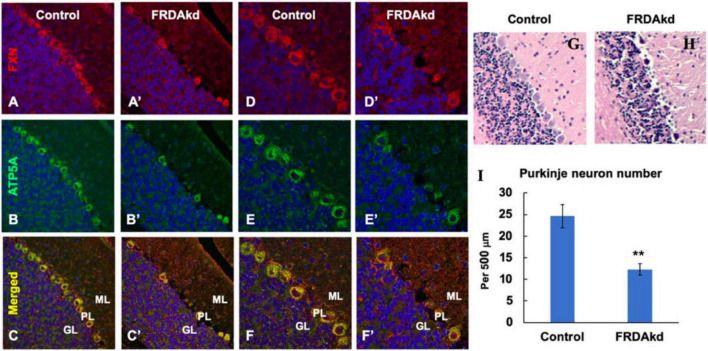
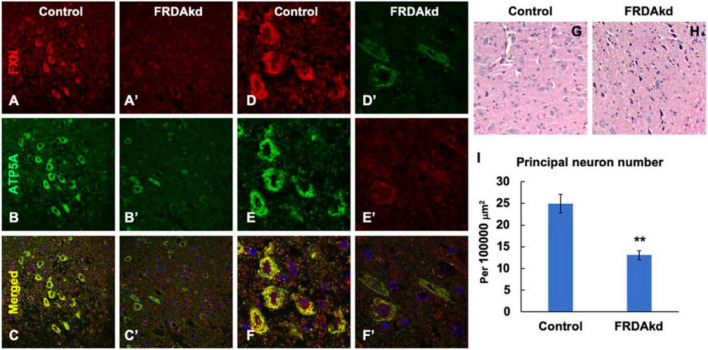
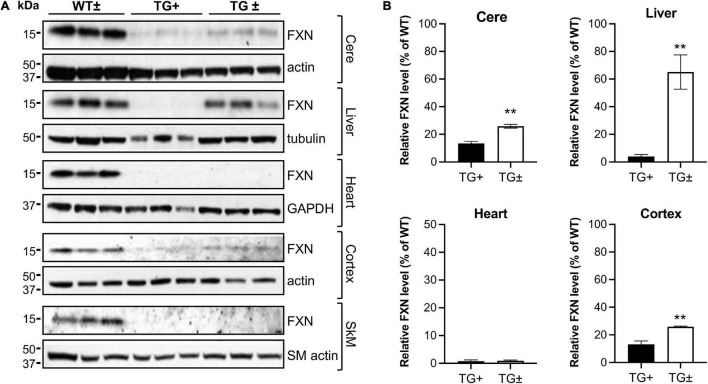
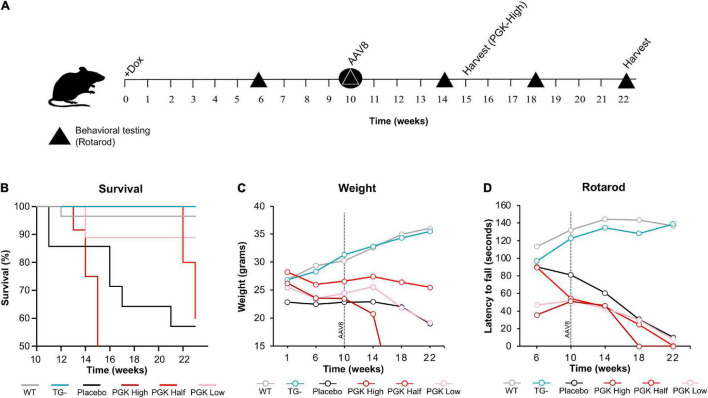
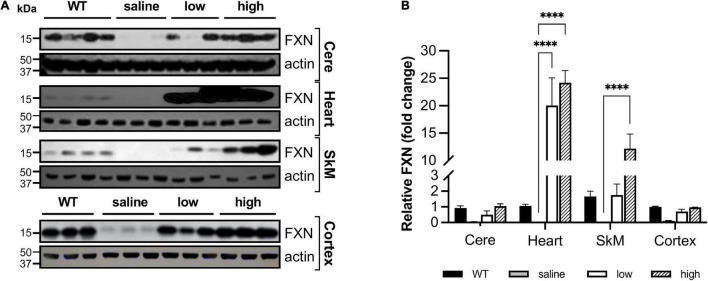
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative disorder caused by deficiency of the mitochondrial protein frataxin. Lack of frataxin causes neuronal loss in various areas of the CNS and PNS. In particular, cerebellar neuropathology in FRDA patients includes loss of large principal neurons and synaptic terminals in the dentate nucleus (DN), and previous studies have demonstrated early synaptic deficits in the Knockin-Knockout mouse model of FRDA. However, the exact correlation of frataxin deficiency with cerebellar neuropathology remains unclear. Here we report that doxycycline-induced frataxin knockdown in a mouse model of FRDA (FRDAkd) leads to synaptic cerebellar degeneration that can be partially reversed by AAV8-mediated frataxin restoration. Loss of cerebellar Purkinje neurons and large DN principal neurons are observed in the FRDAkd mouse cerebellum. Levels of the climbing fiber-specific glutamatergic synaptic marker VGLUT2 decline starting at 4 weeks after dox induction, whereas levels of the parallel fiber-specific synaptic marker VGLUT1 are reduced by 18-weeks. These findings suggest initial selective degeneration of climbing fiber synapses followed by loss of parallel fiber synapses. The GABAergic synaptic marker GAD65 progressively declined during dox induction in FRDAkd mice, while GAD67 levels remained unaltered, suggesting specific roles for frataxin in maintaining cerebellar synaptic integrity and function during adulthood. Expression of frataxin following AAV8-mediated gene transfer partially restored VGLUT1/2 levels. Taken together, our findings show that frataxin knockdown leads to cerebellar degeneration in the FRDAkd mouse model, suggesting that frataxin helps maintain cerebellar structure and function.
弗里德赖希共济失调(FRDA)是一种常染色体隐性神经退行性疾病,由线粒体蛋白酵母氨酸缺乏引起。酵母氨酸缺乏导致中枢神经系统和外周神经系统各区域的神经元丧失。特别是,FRDA患者的小脑神经病理学包括齿状核(DN)中大的主要神经元和突触终末的丧失,并且先前的研究已经在FRDA的敲入-敲除小鼠模型中证明了早期突触缺陷。然而,酵母氨酸缺乏与小脑神经病理学的确切相关性仍不清楚。在此我们报告,在FRDA小鼠模型(FRDAkd)中强力霉素诱导的酵母氨酸敲低导致小脑突触变性,其可被AAV8介导的酵母氨酸恢复部分逆转。在FRDAkd小鼠小脑中观察到小脑浦肯野神经元和大的DN主要神经元的丧失。从强力霉素诱导后4周开始,攀爬纤维特异性谷氨酸能突触标记物VGLUT2的水平下降,而平行纤维特异性突触标记物VGLUT1的水平在18周时降低。这些发现表明攀爬纤维突触最初选择性变性,随后平行纤维突触丧失。在FRDAkd小鼠的强力霉素诱导期间,GABA能突触标记物GAD65逐渐下降,而GAD67水平保持不变,这表明酵母氨酸在成年期维持小脑突触完整性和功能中具有特定作用。AAV8介导的基因转移后酵母氨酸的表达部分恢复了VGLUT1/2水平。综上所述,我们的发现表明酵母氨酸敲低导致FRDAkd小鼠模型中的小脑变性,提示酵母氨酸有助于维持小脑结构和功能。