Jiang Peijia, Chipurupalli Sandhya, Yoo Byong Hoon, Liu Xiaoyang, Rosen Kirill V
Departments of Pediatrics & Biochemistry and Molecular Biology, Dalhousie University, Halifax, NS, Canada.
Cell Death Dis. 2025 Feb 20;16(1):118. doi: 10.1038/s41419-025-07436-z.
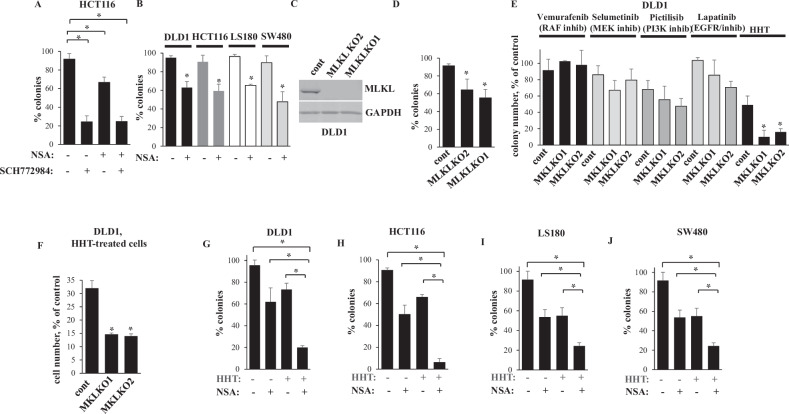
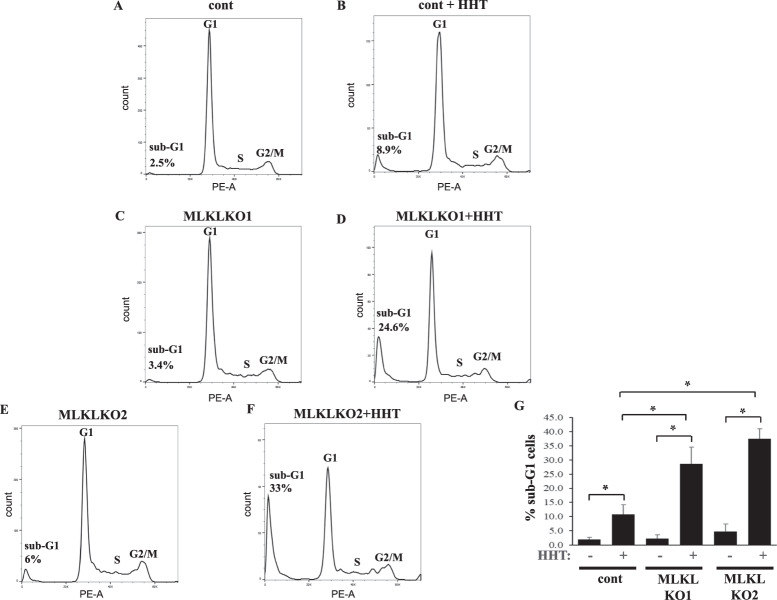
Mortality from colorectal cancer (CRC) is significant, and novel CRC therapies are needed. A pseudokinase MLKL typically executes necroptotic cell death, and MLKL inactivation protects cells from such death. However, we found unexpectedly that MLKL gene knockout enhanced CRC cell death caused by a protein synthesis inhibitor homoharringtonine used for chronic myeloid leukemia treatment. In an effort to explain this finding, we observed that MLKL gene knockout reduces the basal CRC cell autophagy and renders such autophagy critically dependent on the presence of VPS37A, a component of the ESCRT-I complex. We further found that the reason why homoharringtonine enhances CRC cell death caused by MLKL gene knockout is that homoharringtonine activates p38 MAP kinase and thereby prevents VPS37A from supporting autophagy in MLKL-deficient cells. We observed that the resulting inhibition of the basal autophagy in CRC cells triggers their parthanatos, a cell death type driven by poly(ADP-ribose) polymerase hyperactivation. Finally, we discovered that a pharmacological MLKL inhibitor necrosulfonamide strongly cooperates with homoharringtonine in suppressing CRC cell tumorigenicity in mice. Thus, while MLKL promotes cell death during necroptosis, MLKL supports the basal autophagy in CRC cells and thereby protects them from death. MLKL inactivation reduces such autophagy and renders the cells sensitive to autophagy inhibitors, such as homoharringtonine. Hence, MLKL inhibition creates a therapeutic vulnerability that could be utilized for CRC treatment.
结直肠癌(CRC)的死亡率很高,因此需要新的CRC治疗方法。伪激酶混合谱系激酶结构域样蛋白(MLKL)通常执行坏死性凋亡细胞死亡,而MLKL失活可保护细胞免于这种死亡。然而,我们意外地发现,MLKL基因敲除增强了由用于慢性粒细胞白血病治疗的蛋白质合成抑制剂高三尖杉酯碱引起的CRC细胞死亡。为了解释这一发现,我们观察到MLKL基因敲除降低了基础CRC细胞自噬,并使这种自噬严重依赖于内体分选转运复合体I(ESCRT-I)复合物的一个组分VPS37A的存在。我们进一步发现,高三尖杉酯碱增强由MLKL基因敲除引起的CRC细胞死亡的原因是,高三尖杉酯碱激活p38丝裂原活化蛋白激酶(p38 MAP激酶),从而阻止VPS37A在MLKL缺陷细胞中支持自噬。我们观察到,由此导致的CRC细胞基础自噬的抑制触发了它们的细胞焦亡,这是一种由聚(ADP-核糖)聚合酶过度激活驱动的细胞死亡类型。最后,我们发现一种MLKL的药理学抑制剂坏死磺酰胺在抑制小鼠CRC细胞致瘤性方面与高三尖杉酯碱强烈协同作用。因此,虽然MLKL在坏死性凋亡过程中促进细胞死亡,但MLKL支持CRC细胞的基础自噬,从而保护它们免于死亡。MLKL失活会减少这种自噬,并使细胞对自噬抑制剂(如高三尖杉酯碱)敏感。因此,抑制MLKL会产生一种可用于CRC治疗的治疗脆弱性。