Arthur Timothy D, Nguyen Jennifer P, Henson Benjamin A, D'Antonio-Chronowska Agnieszka, Jaureguy Jeffrey, Silva Nayara, Panopoulos Athanasia D, Izpisua Belmonte Juan Carlos, D'Antonio Matteo, McVicker Graham, Frazer Kelly A
Biomedical Sciences Program, University of California, San Diego, La Jolla, CA 92093, USA; Department of Biomedical Informatics, University of California, San Diego, La Jolla, CA 92093, USA.
Department of Biomedical Informatics, University of California, San Diego, La Jolla, CA 92093, USA; Bioinformatics and Systems Biology Graduate Program, University of California, San Diego, La Jolla, CA 92093, USA.
Cell Genom. 2025 Mar 12;5(3):100775. doi: 10.1016/j.xgen.2025.100775. Epub 2025 Feb 21.
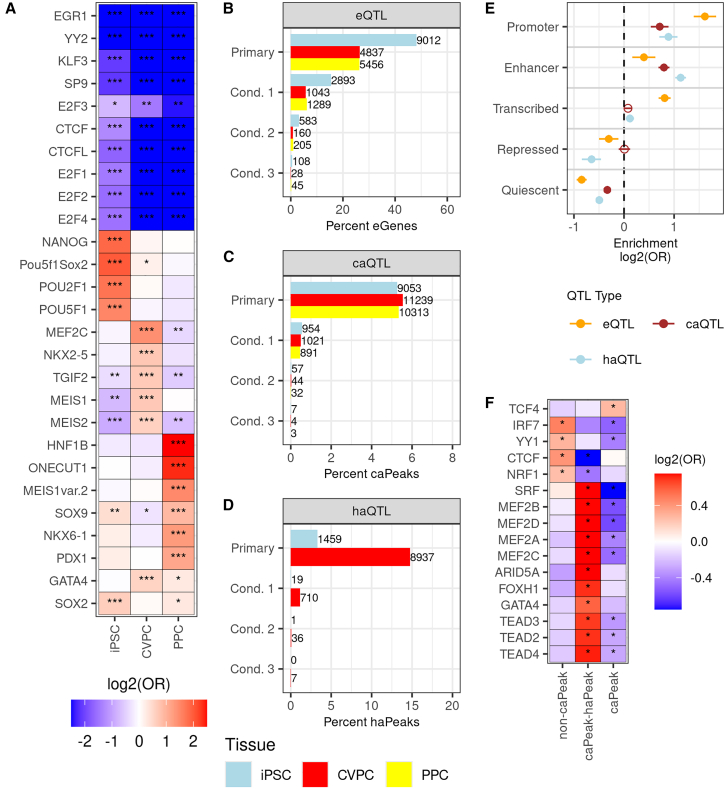
Most GWAS loci are presumed to affect gene regulation; however, only ∼43% colocalize with expression quantitative trait loci (eQTLs). To address this colocalization gap, we map eQTLs, chromatin accessibility QTLs (caQTLs), and histone acetylation QTLs (haQTLs) using molecular samples from three early developmental-like tissues. Through colocalization, we annotate 10.4% (n = 540) of GWAS loci in 15 traits by QTL phenotype, temporal specificity, and complexity. We show that integration of chromatin QTLs results in a 2.3-fold higher annotation rate of GWAS loci because they capture distal GWAS loci missed by eQTLs, and that 5.4% (n = 13) of GWAS colocalizing eQTLs are early developmental specific. Finally, we utilize the iPSCORE multiomic QTLs to prioritize putative causal variants overlapping transcription factor motifs to elucidate the potential genetic underpinnings of 296 GWAS-QTL colocalizations.
大多数全基因组关联研究(GWAS)位点被认为会影响基因调控;然而,只有约43%的位点与表达数量性状位点(eQTL)共定位。为了解决这种共定位差距,我们使用来自三种早期发育样组织的分子样本绘制了eQTL、染色质可及性数量性状位点(caQTL)和组蛋白乙酰化数量性状位点(haQTL)图谱。通过共定位,我们根据QTL表型、时间特异性和复杂性,对15个性状中10.4%(n = 540)的GWAS位点进行了注释。我们表明,染色质QTL的整合使GWAS位点的注释率提高了2.3倍,因为它们捕获了eQTL遗漏的远端GWAS位点,并且5.4%(n = 13)的GWAS共定位eQTL是早期发育特异性的。最后,我们利用iPSCORE多组学QTL对与转录因子基序重叠的推定因果变异进行优先级排序,以阐明296个GWAS-QTL共定位的潜在遗传基础。