Corral-Serrano Julio C, Vaclavik Veronika, Van de Sompele Stijn, Kaminska Karolina, Jovanovic Katarina, Escher Pascal, Van den Broeck Filip, Cancellieri Francesca, Toulis Vasileios, Leroy Bart P, de Zaeytijd Julie, You Zhixuan, Ottaviani Daniele, Quinodoz Mathieu, Bordeanu Gabriela, Hardcastle Alison J, Coppieters Frauke, Tran Viet H, Cheetham Michael E, Rivolta Carlo, De Baere Elfride
UCL Institute of Ophthalmology, EC1V 9EL London, United Kingdom.
Unité d'oculogénétique, Jules Gonin Eye Hospital, University of Lausanne, 1004 Lausanne, Switzerland.
Hum Mol Genet. 2025 Apr 17;34(9):821-834. doi: 10.1093/hmg/ddaf029.
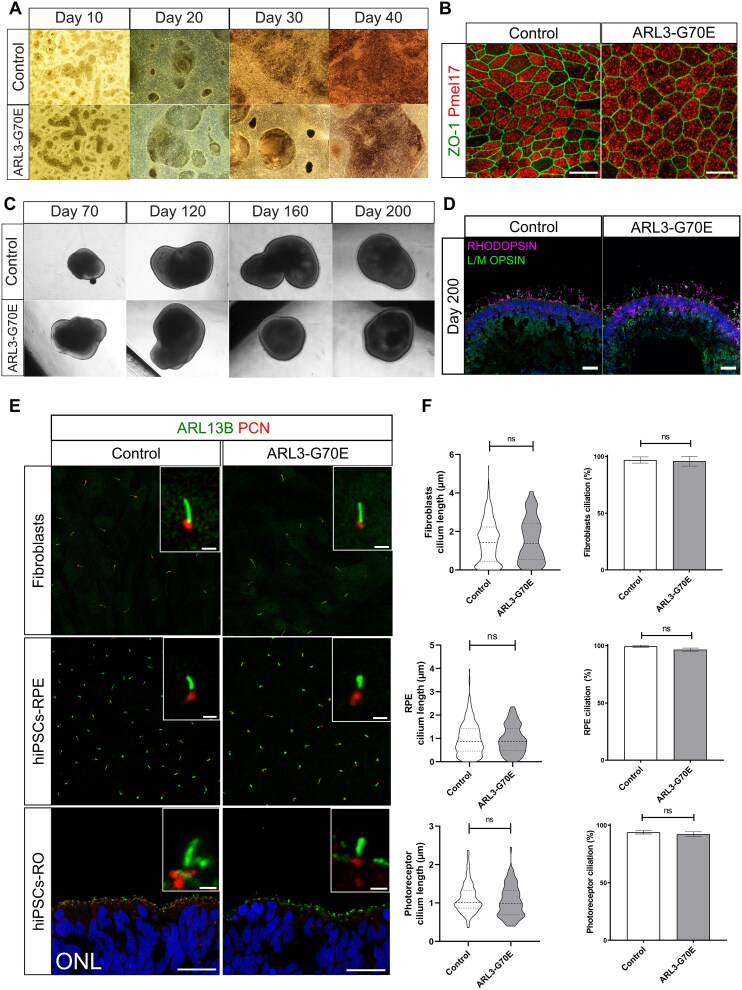
Inherited retinal dystrophies (IRDs) are characterized by their high clinical and genetic heterogeneity. Despite significant advances in the identification of genes associated with IRDs, many individuals and families still have not received a definite molecular diagnosis. Here, we performed clinical examinations and conducted genetic testing in five families with IRD. Whole exome sequencing in the five index cases revealed a heterozygous missense variant, c.209G > A, p.(Gly70Glu) in the ARL3 gene (NM_004311.4). A de novo occurrence was demonstrated in one affected individual and autosomal dominant inheritance in nine affected individuals from four families. Their phenotypes displayed variable expressivity, and ranged from rod-cone to cone-rod dystrophy with photophobia. Human induced pluripotent stem cells (hiPSCs) were generated from dermal fibroblasts from the individual with the de novo ARL3 variant and were differentiated to retinal pigment epithelium cells (RPE) and retinal organoids. Immunofluorescence analyses in these models showed decreased INPP5E localization within the cilia of RPE and connecting cilia of retinal organoids, as well as reduced PDE6⍺ in the organoid outer segments, suggesting that the p.(Gly70Glu) variant causes IRD by defective lipidated protein transport in photoreceptors and/or RPE. This is the first study of ARL3 dysfunction in human retinal cells, highlighting its importance for retinal homeostasis, as well as a variability in the clinical presentation of ARL3-associated IRD.
遗传性视网膜营养不良(IRDs)的特点是具有高度的临床和遗传异质性。尽管在与IRDs相关基因的鉴定方面取得了重大进展,但许多个体和家庭仍未得到明确的分子诊断。在此,我们对五个患有IRD的家庭进行了临床检查和基因检测。对五个先证者进行全外显子测序,发现ARL3基因(NM_004311.4)存在一个杂合错义变异,即c.209G>A,p.(Gly70Glu)。在一名受影响个体中发现该变异为新发突变,在来自四个家庭的九名受影响个体中呈现常染色体显性遗传。他们的表型表现出可变的表达度,范围从视杆 - 视锥营养不良到伴有畏光的视锥 - 视杆营养不良。从携带新发ARL3变异的个体的皮肤成纤维细胞中生成了人诱导多能干细胞(hiPSCs),并将其分化为视网膜色素上皮细胞(RPE)和视网膜类器官。在这些模型中的免疫荧光分析显示,RPE纤毛和视网膜类器官连接纤毛内的INPP5E定位减少,以及类器官外段的PDE6⍺减少,这表明p.(Gly70Glu)变异通过光感受器和/或RPE中脂化蛋白转运缺陷导致IRD。这是首次对人视网膜细胞中ARL3功能障碍进行的研究,突出了其对视网膜内环境稳定的重要性,以及与ARL3相关的IRD临床表现的变异性。