Zou Yurong, Guo Tao, Fu Zhiyuan, Guo Zhongning, Bo Weichen, Yan Dengjie, Wang Qiantao, Zeng Jun, Xu Dingguo, Wang Taijin, Chen Lijuan
State Key Laboratory of Biotherapy and Collaborative Innovation Center of Biotherapy, West China Hospital, Sichuan University, Chengdu, China.
Key Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry and Sichuan Province, West China School of Pharmacy, Sichuan University, Chengdu, China.
Commun Biol. 2025 Mar 12;8(1):422. doi: 10.1038/s42003-025-07840-3.
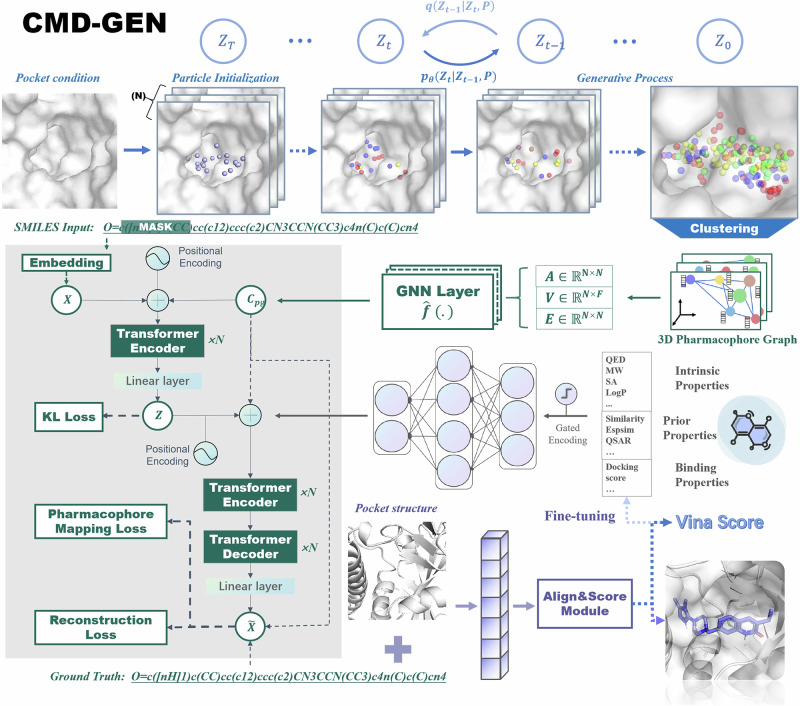
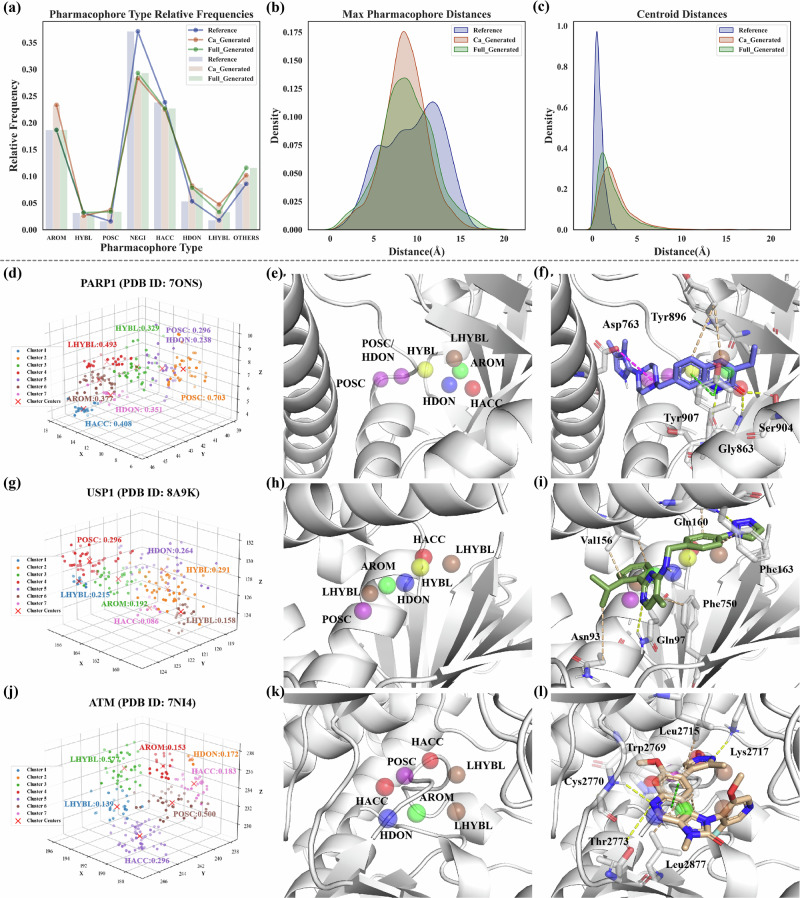
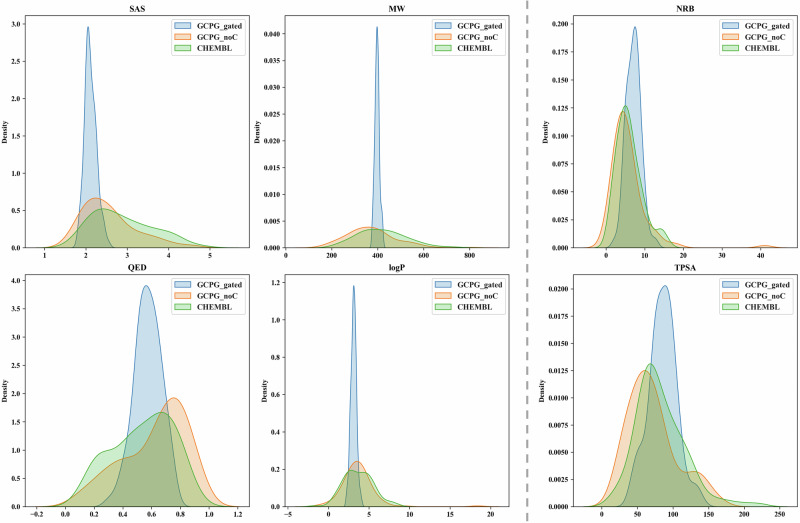
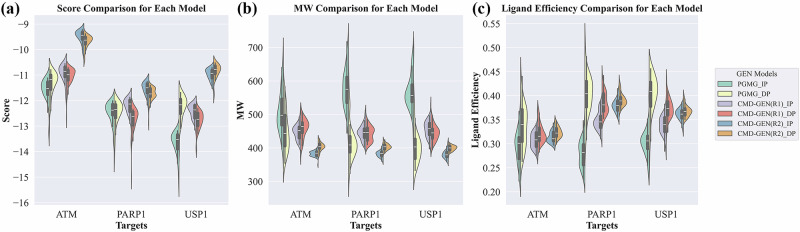
Structure-based drug design aims to create active compounds with favorable properties by analyzing target structures. Recently, deep generative models have facilitated structure-specific molecular generation. However, many methods are limited by inadequate pharmaceutical data, resulting in suboptimal molecular properties and unstable conformations. Additionally, these approaches often overlook binding pocket interactions and struggle with selective inhibitor design. To address these challenges, we developed a framework called Coarse-grained and Multi-dimensional Data-driven molecular generation (CMD-GEN). CMD-GEN bridges ligand-protein complexes with drug-like molecules by utilizing coarse-grained pharmacophore points sampled from diffusion model, enriching training data. Through a hierarchical architecture, it decomposes three-dimensional molecule generation within the pocket into pharmacophore point sampling, chemical structure generation, and conformation alignment, mitigating instability issues. CMD-GEN outperforms other methods in benchmark tests and controls drug-likeness effectively. Furthermore, CMD-GEN excels in cases across three synthetic lethal targets, and wet-lab validation with PARP1/2 inhibitors confirms its potential in selective inhibitor design.
基于结构的药物设计旨在通过分析靶点结构来创建具有良好性质的活性化合物。最近,深度生成模型促进了特定结构的分子生成。然而,许多方法受到药物数据不足的限制,导致分子性质次优和构象不稳定。此外,这些方法常常忽视结合口袋相互作用,并且在选择性抑制剂设计方面存在困难。为了应对这些挑战,我们开发了一个名为粗粒度和多维度数据驱动的分子生成(CMD-GEN)的框架。CMD-GEN通过利用从扩散模型中采样的粗粒度药效团点,将配体-蛋白质复合物与类药物分子联系起来,丰富了训练数据。通过分层架构,它将口袋内的三维分子生成分解为药效团点采样、化学结构生成和构象比对,减轻了不稳定性问题。CMD-GEN在基准测试中优于其他方法,并有效地控制了类药物性质。此外,CMD-GEN在三个合成致死靶点的案例中表现出色,并且用PARP1/2抑制剂进行的湿实验室验证证实了其在选择性抑制剂设计中的潜力。