School of Computer Science and Engineering, Central South University, Changsha, 410083, China.
Xiangya School of Pharmaceutical Sciences, Central South University, Changsha, 410008, China.
Nat Commun. 2023 Oct 6;14(1):6234. doi: 10.1038/s41467-023-41454-9.
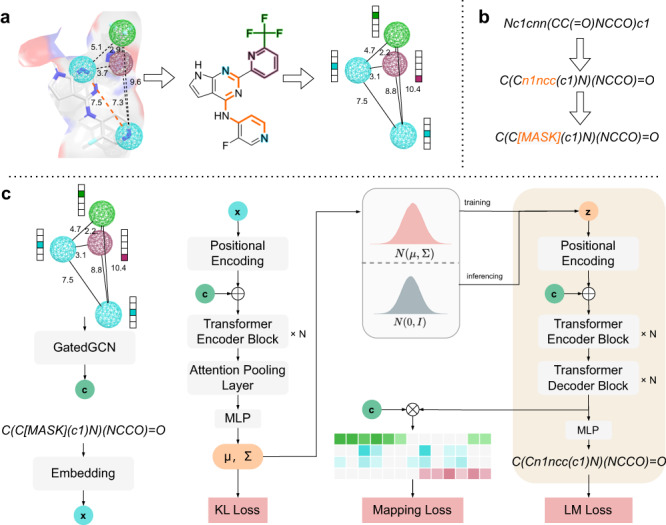
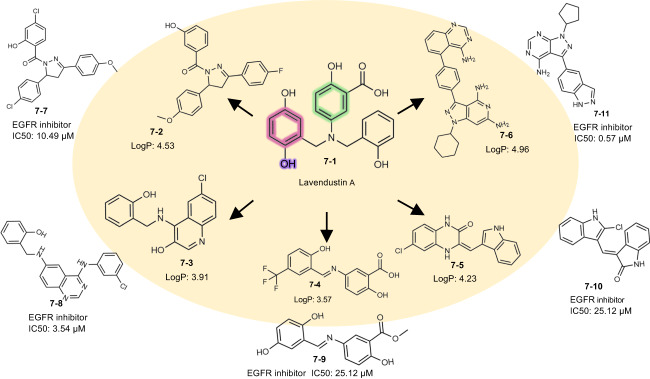
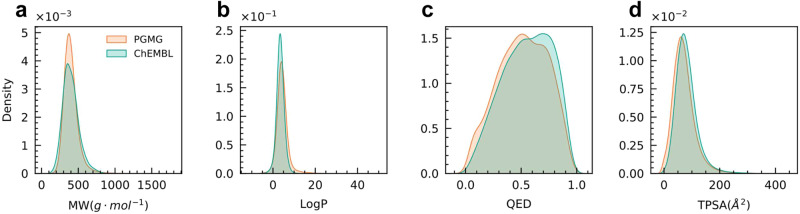
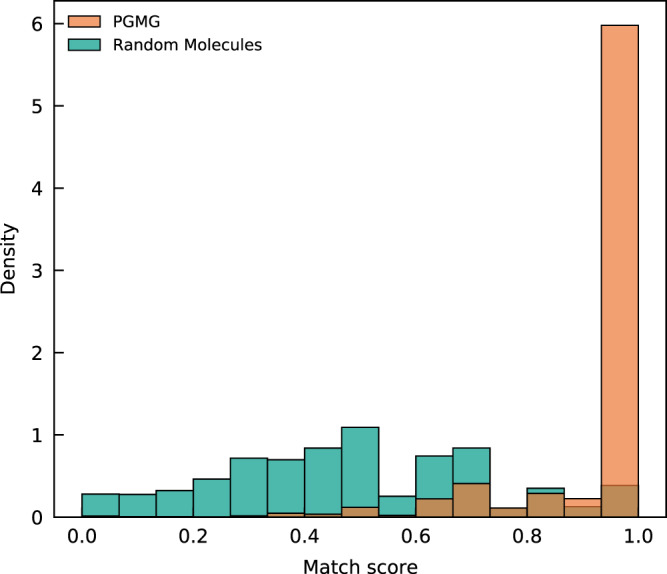
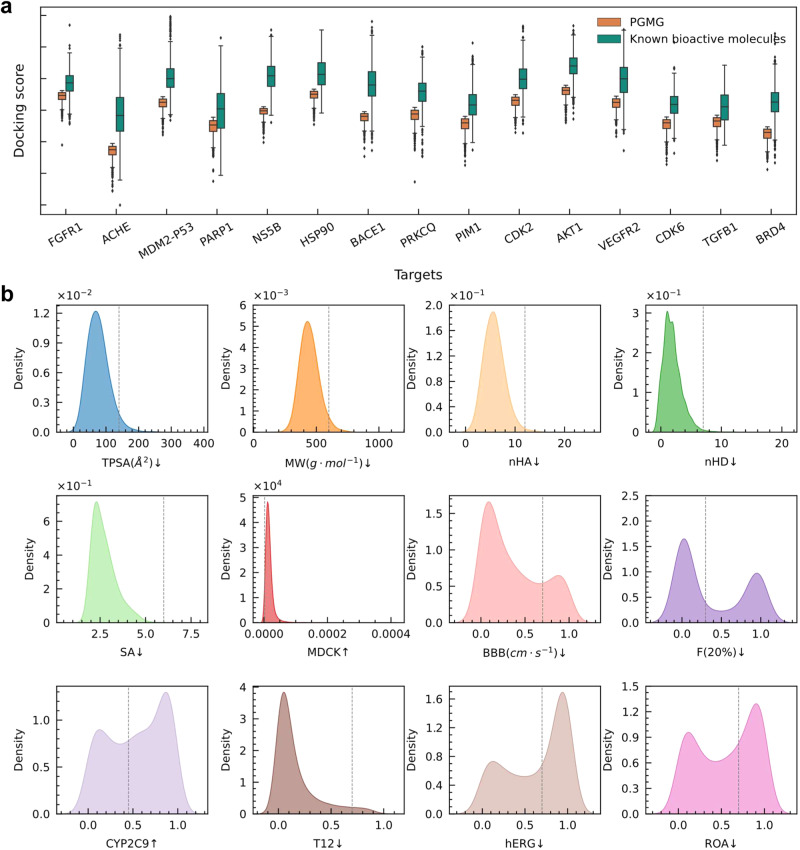
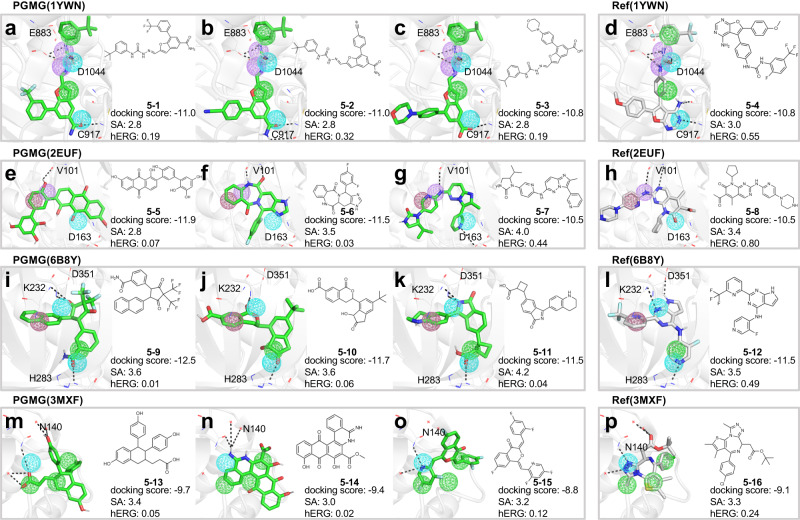
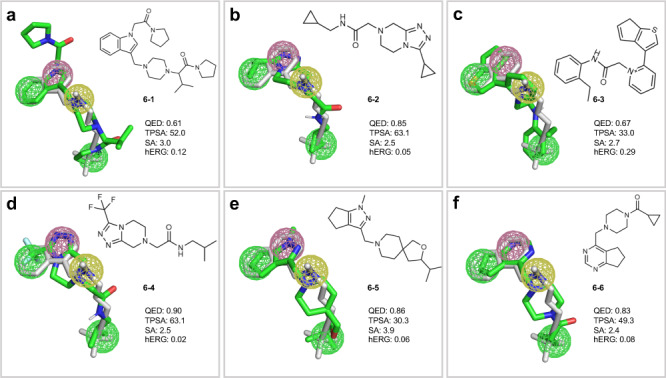
The rational design of novel molecules with the desired bioactivity is a critical but challenging task in drug discovery, especially when treating a novel target family or understudied targets. We propose a Pharmacophore-Guided deep learning approach for bioactive Molecule Generation (PGMG). Through the guidance of pharmacophore, PGMG provides a flexible strategy for generating bioactive molecules. PGMG uses a graph neural network to encode spatially distributed chemical features and a transformer decoder to generate molecules. A latent variable is introduced to solve the many-to-many mapping between pharmacophores and molecules to improve the diversity of the generated molecules. Compared to existing methods, PGMG generates molecules with strong docking affinities and high scores of validity, uniqueness, and novelty. In the case studies, we use PGMG in a ligand-based and structure-based drug de novo design. Overall, the flexibility and effectiveness make PGMG a useful tool to accelerate the drug discovery process.
设计具有预期生物活性的新型分子是药物发现中的一项关键但具有挑战性的任务,尤其是在针对新型靶标家族或研究较少的靶标时。我们提出了一种基于药效团的深度学习方法用于生物活性分子生成(PGMG)。通过药效团的指导,PGMG 为生成生物活性分子提供了一种灵活的策略。PGMG 使用图神经网络来编码空间分布的化学特征,并使用转换器解码器来生成分子。引入了一个潜在变量来解决药效团和分子之间的多对多映射问题,以提高生成分子的多样性。与现有方法相比,PGMG 生成的分子具有较强的对接亲和力和较高的有效性、独特性和新颖性得分。在案例研究中,我们在基于配体和基于结构的药物从头设计中使用了 PGMG。总体而言,其灵活性和有效性使 PGMG 成为加速药物发现过程的有用工具。