Chen Yangyang, Wang Zixu, Wang Lei, Wang Jianmin, Li Pengyong, Cao Dongsheng, Zeng Xiangxiang, Ye Xiucai, Sakurai Tetsuya
Department of Computer Science, University of Tsukuba, Tsukuba, 3058577, Japan.
Xiangya School of Pharmaceutical Sciences, Central South University, Changsha, 410013, Hunan, China.
J Cheminform. 2023 Mar 28;15(1):38. doi: 10.1186/s13321-023-00702-2.
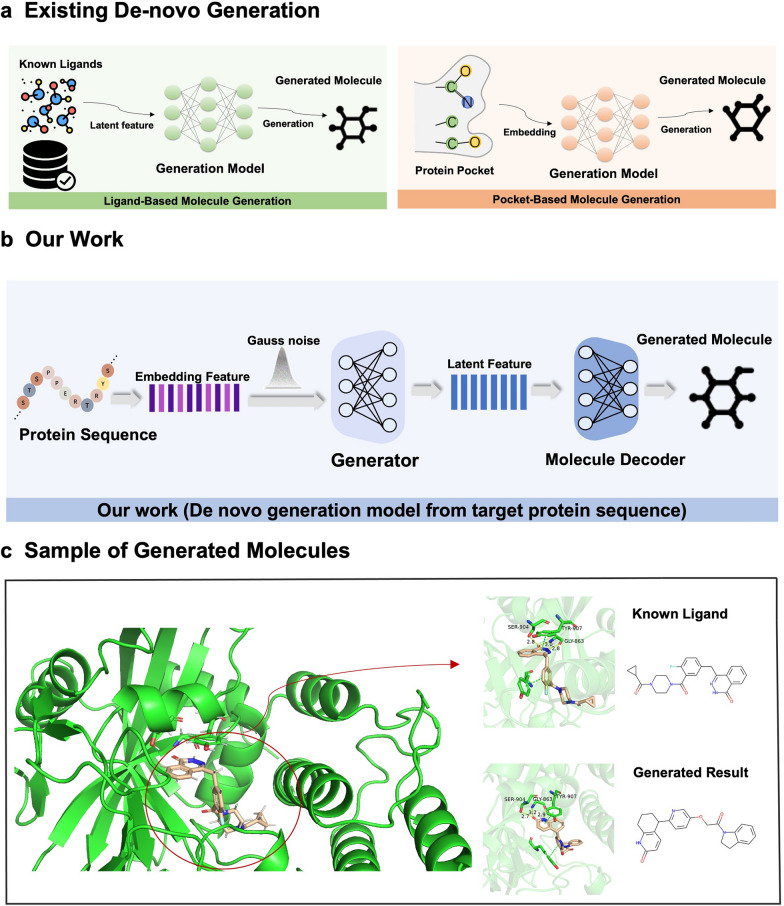
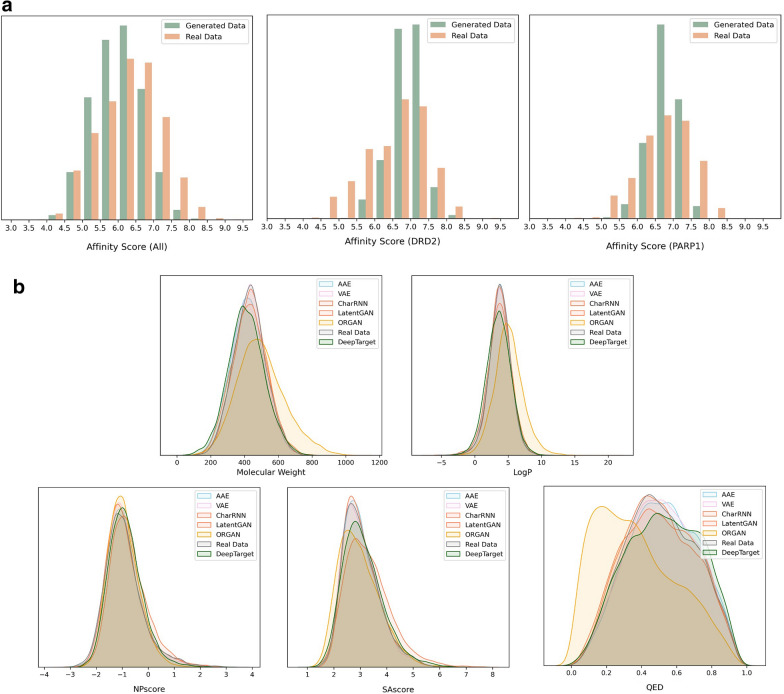
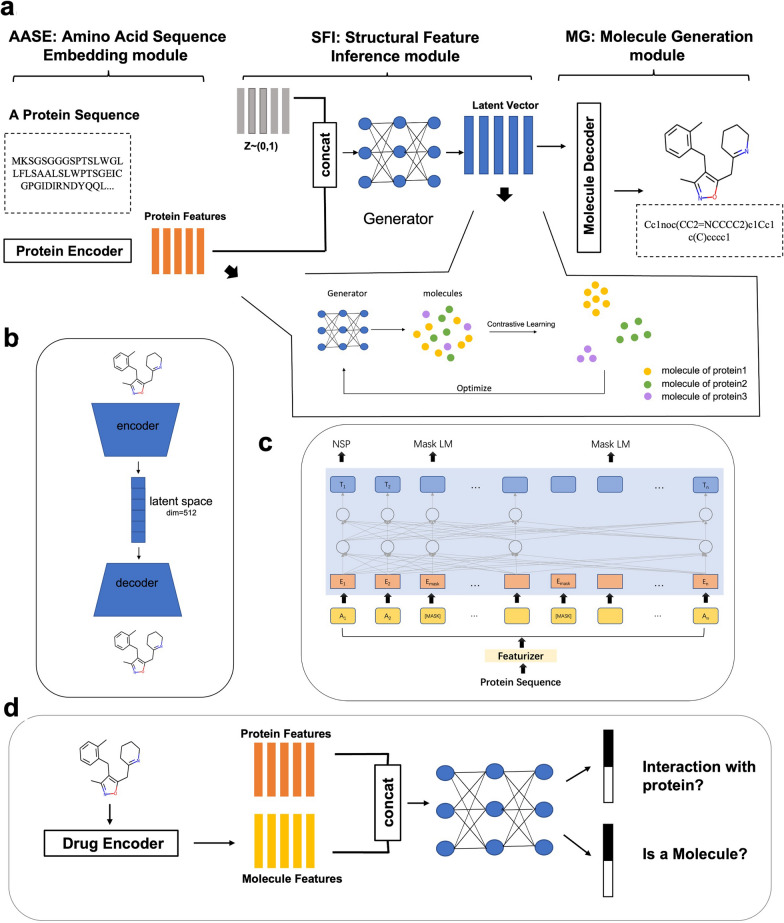
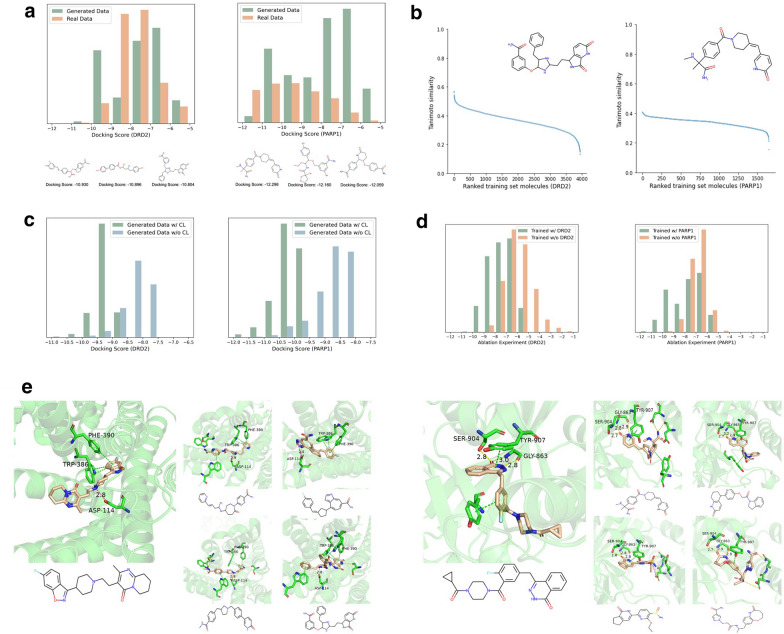
Drug discovery for a protein target is a laborious and costly process. Deep learning (DL) methods have been applied to drug discovery and successfully generated novel molecular structures, and they can substantially reduce development time and costs. However, most of them rely on prior knowledge, either by drawing on the structure and properties of known molecules to generate similar candidate molecules or extracting information on the binding sites of protein pockets to obtain molecules that can bind to them. In this paper, DeepTarget, an end-to-end DL model, was proposed to generate novel molecules solely relying on the amino acid sequence of the target protein to reduce the heavy reliance on prior knowledge. DeepTarget includes three modules: Amino Acid Sequence Embedding (AASE), Structural Feature Inference (SFI), and Molecule Generation (MG). AASE generates embeddings from the amino acid sequence of the target protein. SFI inferences the potential structural features of the synthesized molecule, and MG seeks to construct the eventual molecule. The validity of the generated molecules was demonstrated by a benchmark platform of molecular generation models. The interaction between the generated molecules and the target proteins was also verified on the basis of two metrics, drug-target affinity and molecular docking. The results of the experiments indicated the efficacy of the model for direct molecule generation solely conditioned on amino acid sequence.
针对蛋白质靶点的药物发现是一个费力且成本高昂的过程。深度学习(DL)方法已被应用于药物发现,并成功生成了新的分子结构,而且它们可以大幅减少开发时间和成本。然而,其中大多数方法依赖于先验知识,要么借鉴已知分子的结构和性质来生成相似的候选分子,要么提取蛋白质口袋结合位点的信息以获得能够与之结合的分子。在本文中,提出了一种端到端的深度学习模型DeepTarget,它仅依靠目标蛋白质的氨基酸序列来生成新分子,以减少对先验知识的严重依赖。DeepTarget包括三个模块:氨基酸序列嵌入(AASE)、结构特征推断(SFI)和分子生成(MG)。AASE从目标蛋白质的氨基酸序列生成嵌入。SFI推断合成分子的潜在结构特征,而MG则试图构建最终的分子。通过一个分子生成模型的基准平台证明了所生成分子的有效性。还基于药物-靶点亲和力和分子对接这两个指标验证了所生成分子与目标蛋白质之间的相互作用。实验结果表明了该模型仅根据氨基酸序列直接生成分子的有效性。