Koudelka Tomas, Bassot Claudio, Piazza Ilaria
Max-Delbrück-Center for Molecular Medicine in the Helmholtz Association (MDC), Berlin, Germany.
Max-Delbrück-Center for Molecular Medicine in the Helmholtz Association (MDC), Berlin, Germany; SciLifeLab, Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Solna, Sweden.
Mol Cell Proteomics. 2025 Apr;24(4):100945. doi: 10.1016/j.mcpro.2025.100945. Epub 2025 Mar 13.
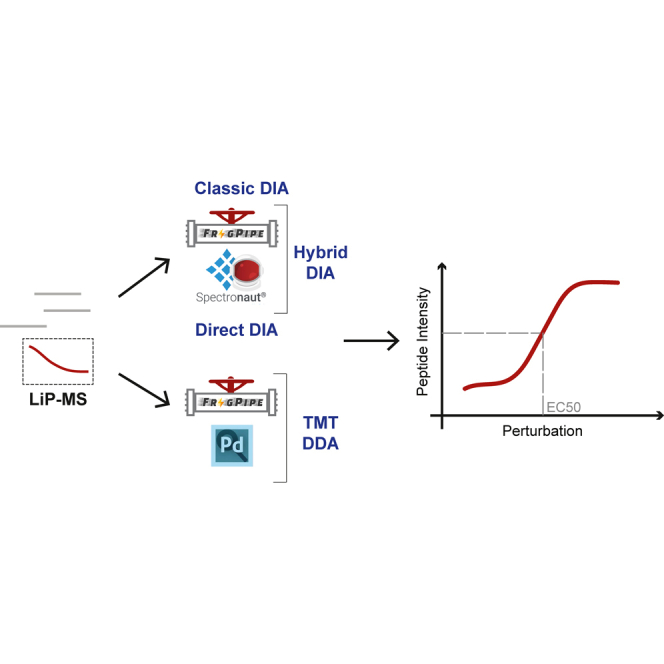
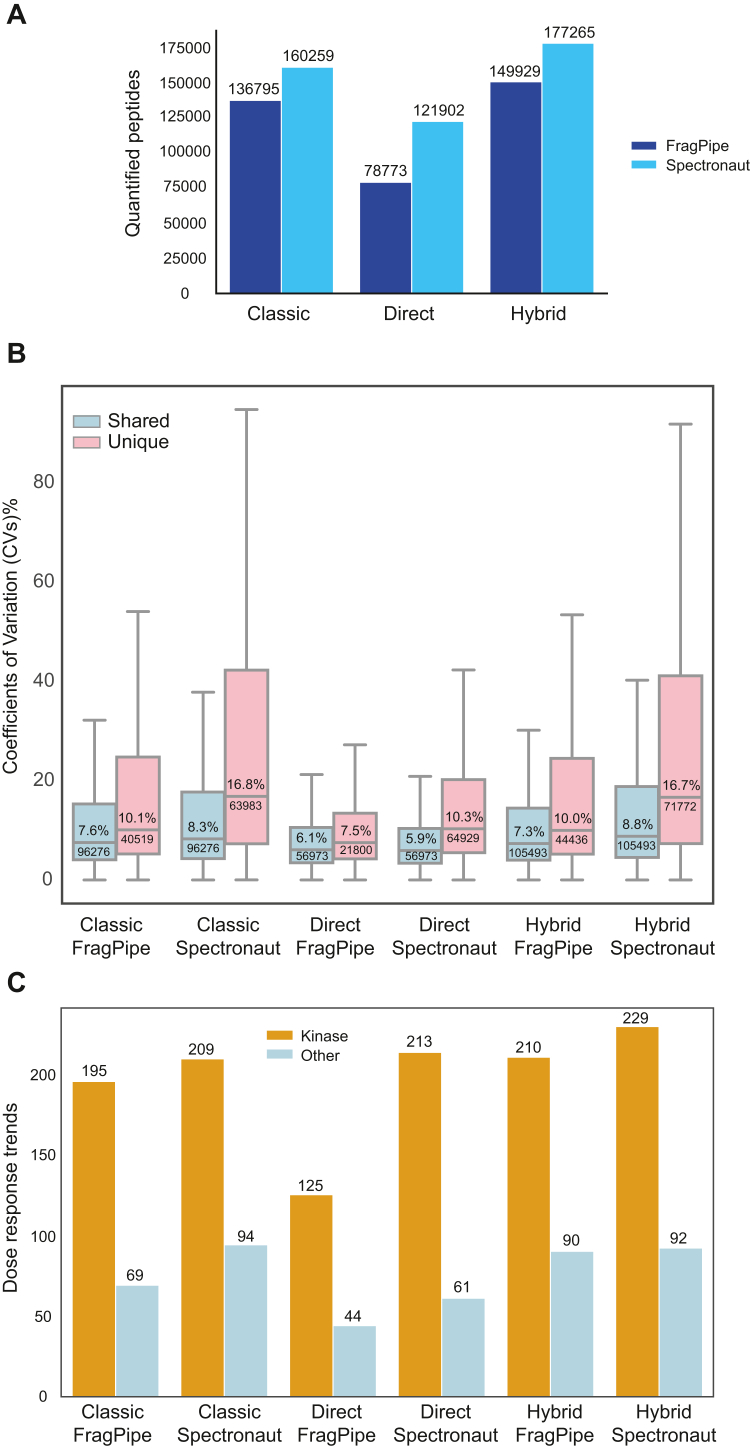
Limited proteolysis coupled with mass spectrometry (LiP-MS) has emerged as a powerful technique for detecting protein structural changes and drug-protein interactions on a proteome-wide scale. However, there is no consensus on the best quantitative proteomics workflow for analyzing LiP-MS data. In this study, we comprehensively benchmarked two major quantification approaches-data-independent acquisition (DIA) and tandem mass tag (TMT) isobaric labeling-in combination with LiP-MS, using a drug-target deconvolution assay as a model system. Our results show that while TMT labeling enabled the quantification of more peptides and proteins with lower coefficients of variation, DIA-MS exhibited greater accuracy in identifying true drug targets and stronger dose-response correlation in peptides of protein targets. Additionally, we evaluated the performance of freely available (FragPipe) versus commercial (Spectronaut) software tools for DIA-MS analysis, revealing that the choice between precision (FragPipe) and sensitivity (Spectronaut) largely depends on the specific experimental context. Our findings underscore the importance of selecting the appropriate LiP-MS quantification strategy based on the study objectives. This work provides valuable guidelines for researchers in structural proteomics and drug discovery, and highlights how advancements in mass spectrometry instrumentation, such as the Astral mass spectrometer, may further improve sensitivity and protein sequence coverage, potentially reducing the need for TMT labeling.
有限蛋白水解结合质谱法(LiP-MS)已成为一种强大的技术,可在蛋白质组范围内检测蛋白质结构变化和药物-蛋白质相互作用。然而,对于分析LiP-MS数据的最佳定量蛋白质组学工作流程尚无共识。在本研究中,我们使用药物-靶点反卷积分析作为模型系统,全面比较了两种主要的定量方法——数据非依赖采集(DIA)和串联质谱标签(TMT)等压标记——与LiP-MS结合的情况。我们的结果表明,虽然TMT标记能够定量更多的肽段和蛋白质,且变异系数较低,但DIA-MS在识别真正的药物靶点方面表现出更高的准确性,在蛋白质靶点的肽段中具有更强的剂量反应相关性。此外,我们评估了用于DIA-MS分析的免费软件工具(FragPipe)与商业软件工具(Spectronaut)的性能,发现精度(FragPipe)和灵敏度(Spectronaut)之间的选择很大程度上取决于具体的实验背景。我们的研究结果强调了根据研究目标选择合适的LiP-MS定量策略的重要性。这项工作为结构蛋白质组学和药物发现领域的研究人员提供了有价值的指导,并突出了质谱仪器的进步,如Astral质谱仪,可能如何进一步提高灵敏度和蛋白质序列覆盖率,从而有可能减少对TMT标记的需求。