Xiao Xu, Ran Zhaohui, Yan Chao, Gu Weihao, Li Zhi
College of Forestry, Guizhou University, Guiyang, 550025, China.
BMC Plant Biol. 2025 Apr 5;25(1):435. doi: 10.1186/s12870-025-06461-6.
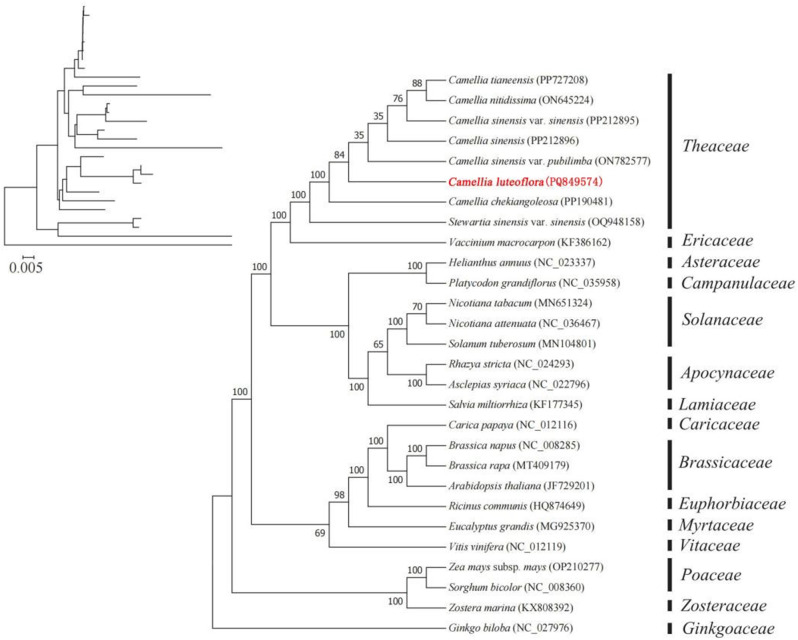
Camellia luteoflora Y.K. Li ex Hung T. Chang & F.A. Zeng belongs to the Camellia L. genus (Theaceae Mirb.). As an endemic, rare, and critically endangered species in China, it holds significant ornamental and economic value, garnering global attention due to its ecological rarity. Despite its conservation importance, genomic investigations on this species remain limited, particularly in organelle genomics, hindering progress in phylogenetic classification and population identification. In this study, we employed high-throughput sequencing to assemble the first complete mitochondrial genome of C. luteoflora and reannotated its chloroplast genome. Through integrated bioinformatics analyses, we systematically characterized the mitochondrial genome's structural organization, gene content, interorganellar DNA transfer, sequence variation, and evolutionary relationships.Key findings revealed a circular mitochondrial genome spanning 587,847 bp with a GC content of 44.63%. The genome harbors70 unique functional genes, including 40 protein-coding genes (PCGs), 27 tRNA genes, and 3 rRNA genes. Notably, 9 PCGs contained 22 intronic regions. Codon usage analysis demonstrated a pronounced A/U bias in synonymous codon selection. Structural features included 506 dispersed repeats and 240 simple sequence repeats. Comparative genomics identified 19 chloroplast-derived transfer events, contributing 29,534 bp (3.77% of total mitochondrial DNA). RNA editing prediction revealed 539 C-to-T conversion events across PCGs. Phylogenetic reconstruction using mitochondrial PCGs positioned C. luteoflora in closest evolutionary proximity to Camellia sinensis var. sinensis. Selection pressure analysis (Ka/Ks ratios < 1 for 11 PCGs) and nucleotide diversity assessment (Pi values: 0-0.00711) indicated strong purifying selection and low sequence divergence.This study provides the first comprehensive mitochondrial genomic resource for C. luteoflora, offering critical insights for germplasm conservation, comparative organelle genomics, phylogenetic resolution, and evolutionary adaptation studies in Camellia species.
金花茶(Camellia luteoflora Y.K. Li ex Hung T. Chang & F.A. Zeng)属于山茶属(Theaceae Mirb.)。作为中国特有的珍稀濒危物种,它具有重要的观赏和经济价值,因其生态稀有性而受到全球关注。尽管其具有保护重要性,但对该物种的基因组研究仍然有限,特别是在细胞器基因组学方面,这阻碍了系统发育分类和种群鉴定的进展。在本研究中,我们采用高通量测序技术组装了金花茶的首个完整线粒体基因组,并对其叶绿体基因组进行了重新注释。通过综合生物信息学分析,我们系统地描述了线粒体基因组的结构组织、基因内容、细胞器间DNA转移、序列变异和进化关系。主要发现揭示了一个环状线粒体基因组,长度为587,847 bp,GC含量为44.63%。该基因组包含70个独特的功能基因,包括40个蛋白质编码基因(PCG)、27个tRNA基因和3个rRNA基因。值得注意的是,9个PCG包含22个内含子区域。密码子使用分析表明,同义密码子选择存在明显的A/U偏好。结构特征包括506个分散重复和240个简单序列重复。比较基因组学确定了19个来自叶绿体的转移事件,贡献了29,534 bp(占线粒体DNA总量的3.77%)。RNA编辑预测揭示了PCG中539个C到T的转换事件。使用线粒体PCG进行的系统发育重建将金花茶定位在与中华变种茶树进化关系最密切的位置。选择压力分析(11个PCG的Ka/Ks比值<1)和核苷酸多样性评估(Pi值:0-0.00711)表明存在强烈的纯化选择和低序列差异。本研究为金花茶提供了首个全面的线粒体基因组资源,为山茶属物种的种质保护、比较细胞器基因组学、系统发育解析和进化适应研究提供了关键见解。