Schlinsog Megan, Sattasathuchana Tosaporn, Xu Peng, Guidez Emilie B, Gilson Michael K, Potter Michael J, Gordon Mark S, Webb Simon P
Department of Chemistry, Iowa State University and Ames National Laboratory, Ames, Iowa 50014, United States.
Department of Chemistry, University of Hawai'i at Manoa, Honolulu, Hawaii 96822, United States.
J Chem Theory Comput. 2025 Apr 22;21(8):4236-4265. doi: 10.1021/acs.jctc.4c01707. Epub 2025 Apr 9.
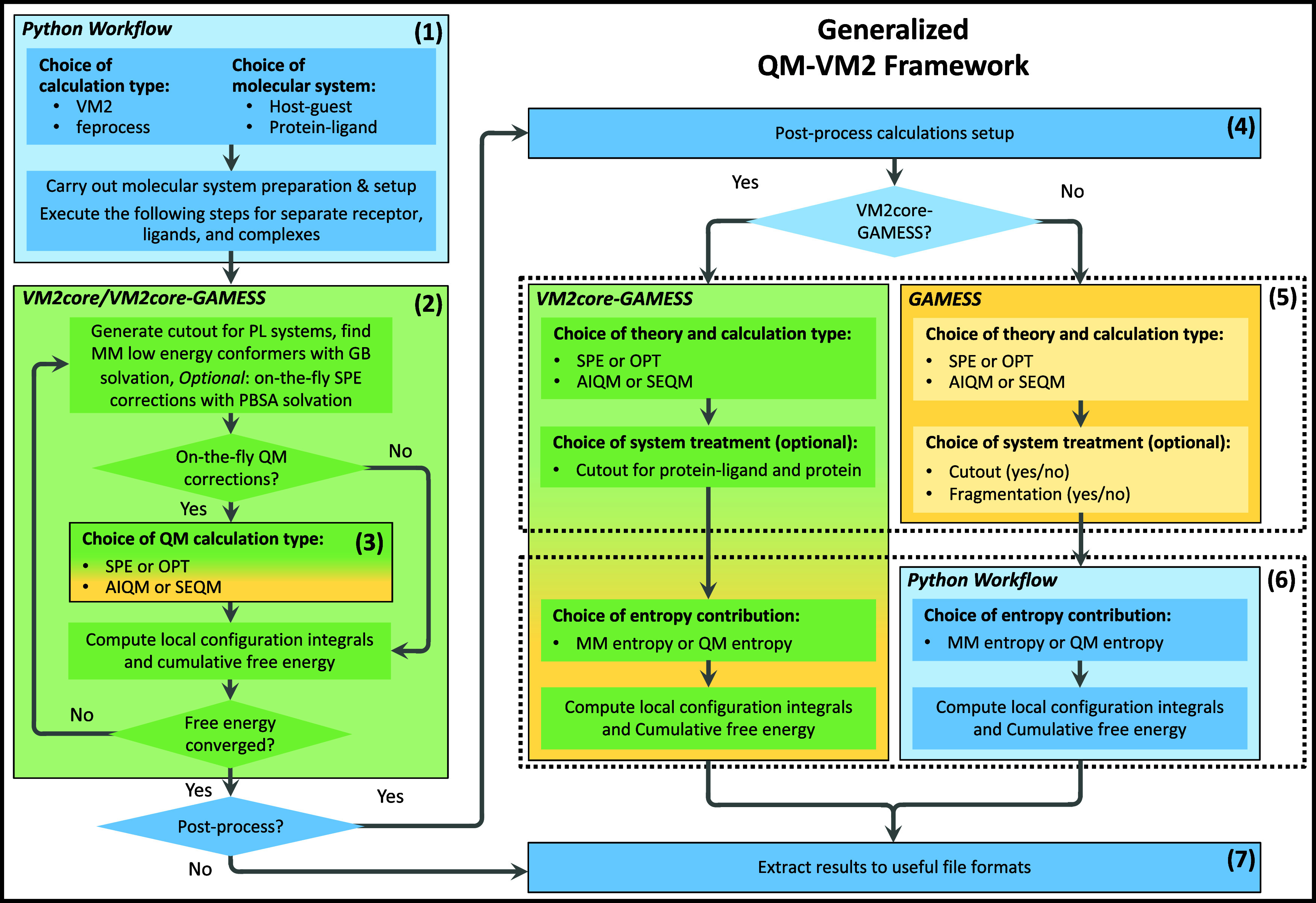
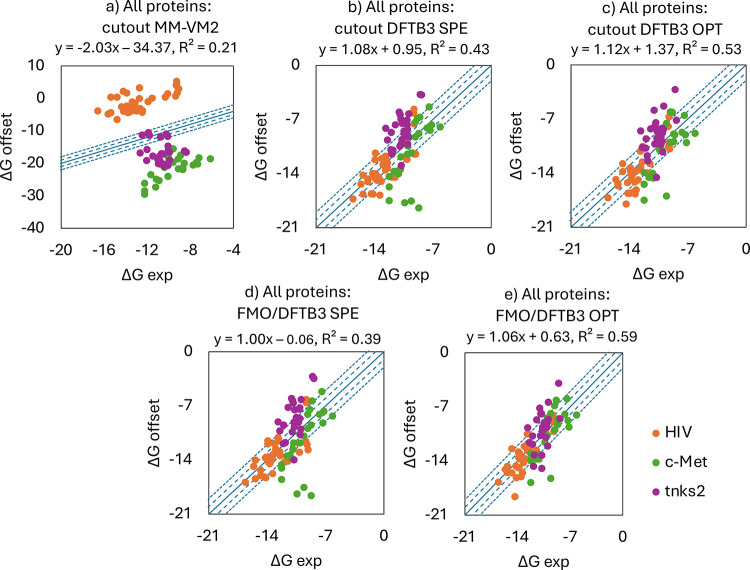
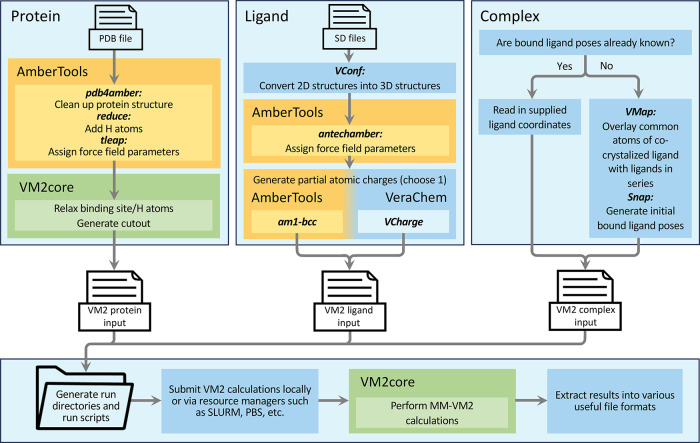
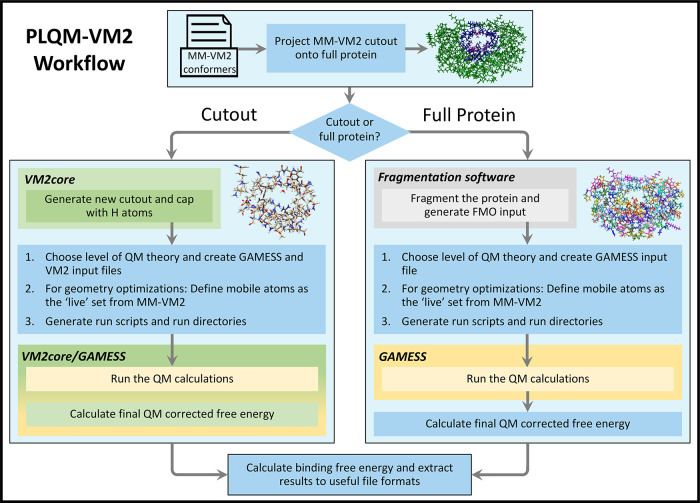
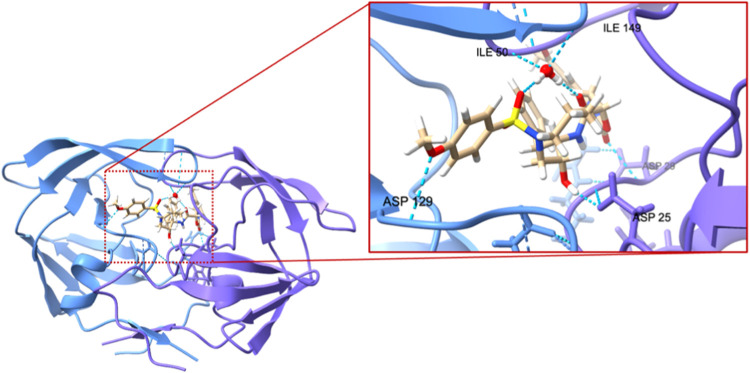
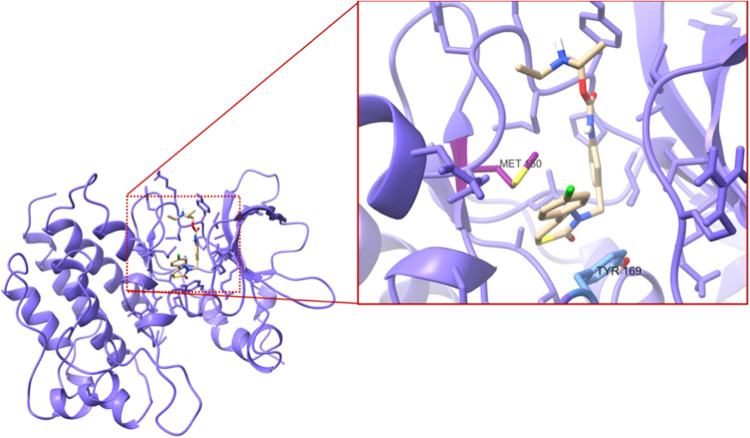
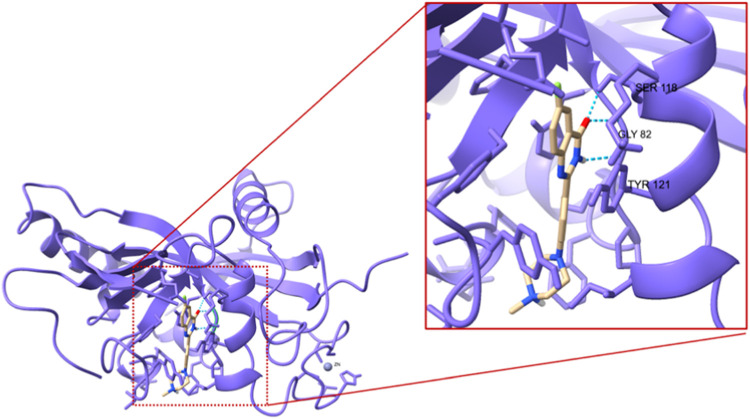
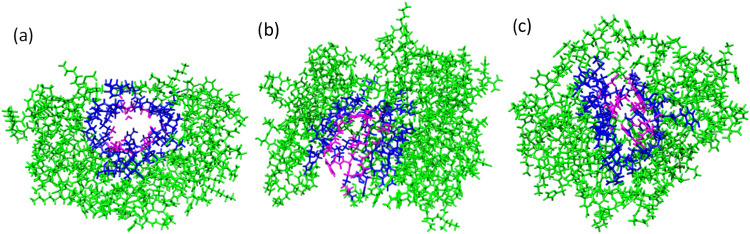
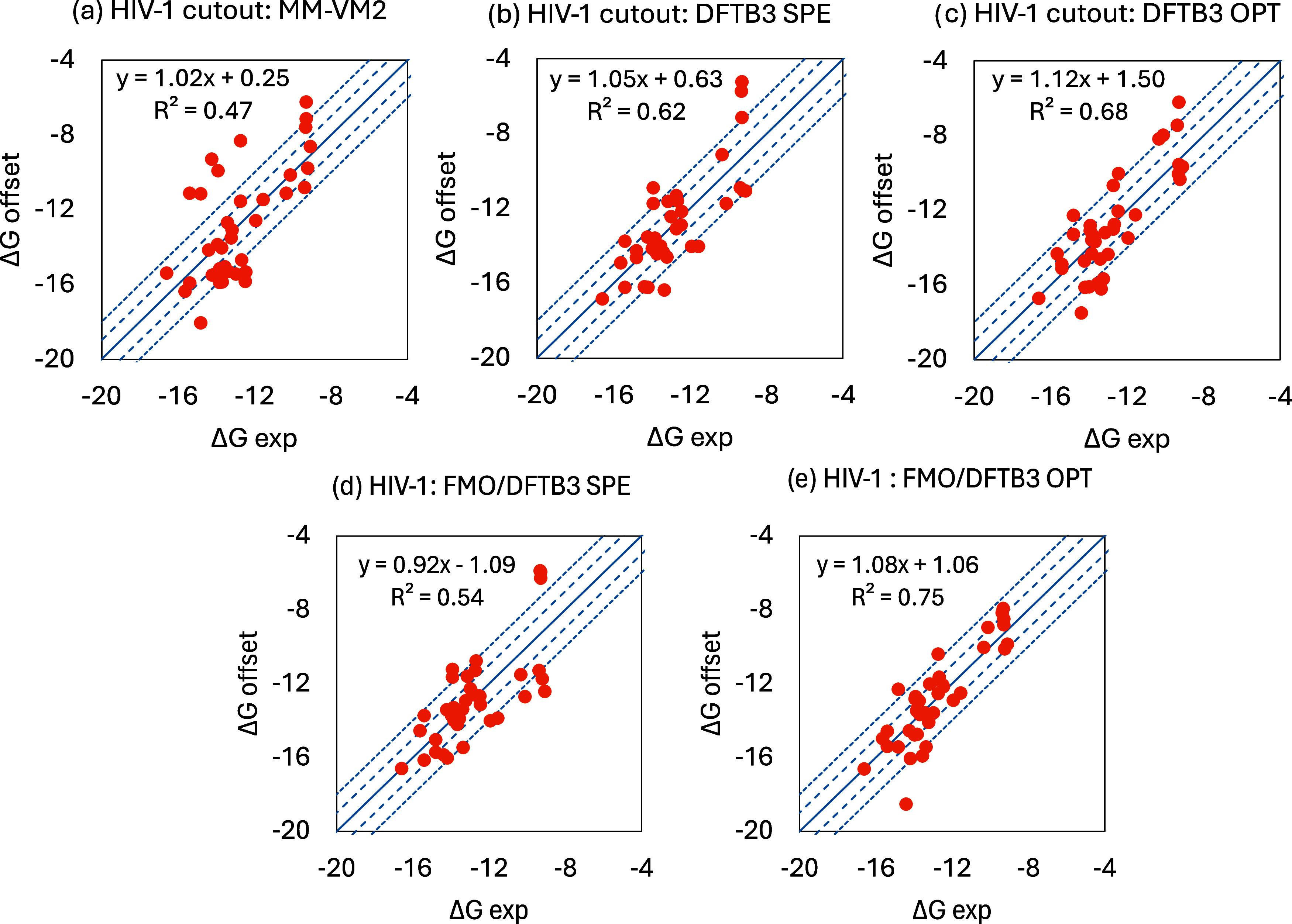
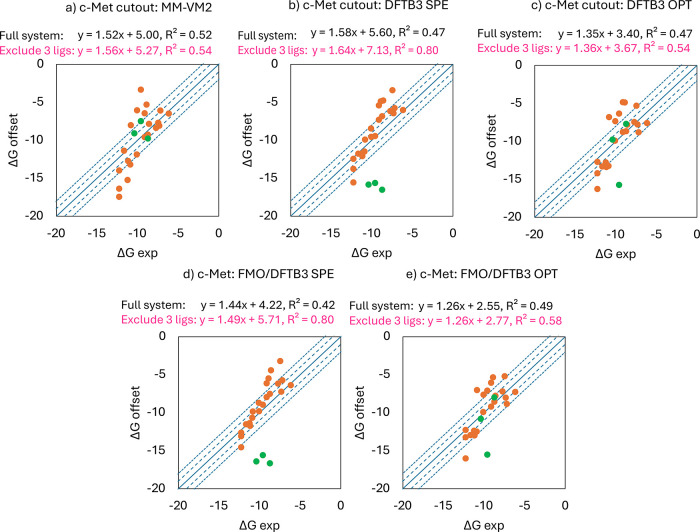
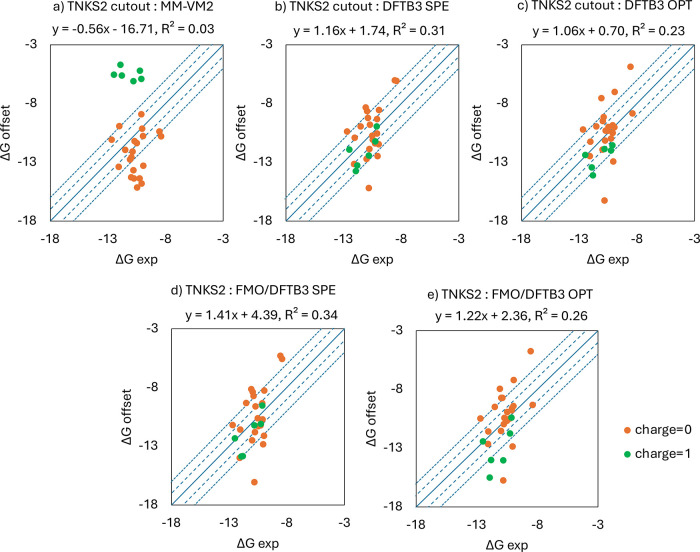
A new method, protein-ligand QM-VM2 (PLQM-VM2), to calculate protein-ligand binding free energies by combining mining minima, a statistical mechanics end-point-based approach, with quantum mechanical potentials is presented. PLQM-VM2 is described in terms of a highly flexible workflow that is initiated from a Protein Data Bank (PDB) file and a chemical structure data file (SD file) containing two-dimensional (2D) or three-dimensional (3D) ligand series coordinates. The workflow utilizes the previously developed molecular mechanics (MM) implementation of the second-generation mining minima method, MM-VM2, to provide ensembles of protein, free ligand, and protein-ligand conformers, which are postprocessed at chosen levels of QM theory, via the quantum chemistry software package GAMESS, to correct MM-based conformer geometries and electronic energies. The corrected energies are used in the calculation of configuration integrals, which on summation over the conformer ensembles give QM-corrected chemical potentials and ultimately QM-corrected binding free energies. In this work, PLQM-VM2 is applied to three benchmark protein-ligand series: HIV-1 protease/38 ligands, c-Met/24 ligands, and TNKS2/27 ligands. QM corrections are carried out at the semiempirical third-order density functional tight-binding level of theory, augmented with dispersion and damping corrections (DFTB3-D3(BJ)H). Bulk solvation effects are accounted for with the conductor-like polarizable continuum model (PCM). DFTB3-D3(BJ)H/PCM single-point energy-only and geometry optimization QM corrections are carried out in conjunction with two different models that address the large computational scaling associated with protein-sized molecular systems. One is a protein cutout model, whereby a set of protein atoms in and around the binding site are carved out, dangling bonds are capped with hydrogens, and only atoms directly in the protein binding site are mobile along with the ligand atoms. The other model is the Fragment Molecular Orbital (FMO) method, which includes the whole protein system but again only allows the binding site and ligand atoms to be mobile. All four of these methodological approaches to QM corrections provide significant improvement over MM-VM2 in terms of rank order and parametric linear correlation with experimentally determined binding affinities. Overall, FMO with geometry optimizations performed the best, but the much cheaper cutout single-point energy approach still provides a good level of accuracy. Furthermore, a clear result is that the PLQM-VM2 calculated binding free energies for the three diverse test systems in this work are, in contrast to those calculated using MM-VM2, directly comparable in energy scale. This suggests a basis for future development of a PLQM-VM2-based multiprotein screening capability to check for off-target activity of ligand series. Benchmark timings on a single compute node (32 CPU cores) for PLQM-VM2 calculation of the chemical potential of a protein-ligand complex range from ca. 30-45 min for the single-point energy approaches to ∼5 h for the cutout approach with geometry optimization and to ∼35 h for the full protein FMO approach with geometry optimization.
本文提出了一种新方法——蛋白质-配体QM-VM2(PLQM-VM2),该方法通过将挖掘极小值(一种基于统计力学端点的方法)与量子力学势相结合来计算蛋白质-配体结合自由能。PLQM-VM2是通过一个高度灵活的工作流程来描述的,该流程从蛋白质数据库(PDB)文件和包含二维(2D)或三维(3D)配体系列坐标的化学结构数据文件(SD文件)开始。该工作流程利用先前开发的第二代挖掘极小值方法MM-VM2的分子力学(MM)实现,来提供蛋白质、游离配体和蛋白质-配体构象异构体的集合,这些构象异构体在选定的量子力学(QM)理论水平上,通过量子化学软件包GAMESS进行后处理,以校正基于MM的构象异构体几何结构和电子能量。校正后的能量用于计算构型积分,对构象异构体集合求和后可得到QM校正的化学势,并最终得到QM校正的结合自由能。在这项工作中,PLQM-VM2应用于三个基准蛋白质-配体系列:HIV-1蛋白酶/38个配体、c-Met/24个配体和TNKS2/27个配体。QM校正是在半经验三阶密度泛函紧束缚理论水平上进行的,并增加了色散和阻尼校正(DFTB3-D3(BJ)H)。采用导体类极化连续介质模型(PCM)考虑整体溶剂化效应。DFTB3-D3(BJ)H/PCM单点能量仅和几何优化QM校正与两种不同模型结合进行,这两种模型解决了与蛋白质大小的分子系统相关的大规模计算缩放问题。一种是蛋白质剪裁模型,即从结合位点及其周围切割出一组蛋白质原子,用氢原子封端悬空键,并且只有直接在蛋白质结合位点的原子与配体原子一起移动。另一种模型是片段分子轨道(FMO)方法,它包括整个蛋白质系统,但同样只允许结合位点和配体原子移动。所有这四种QM校正方法在排序以及与实验测定的结合亲和力的参数线性相关性方面都比MM-VM2有显著改进。总体而言,进行几何优化的FMO表现最佳,但成本低得多的剪裁单点能量方法仍提供了较高的准确度。此外,一个明确的结果是,与使用MM-VM2计算的结果相比,PLQM-VM2为本工作中三个不同测试系统计算的结合自由能在能量尺度上直接可比。这为未来基于PLQM-VM2开发多蛋白筛选能力以检查配体系列的脱靶活性奠定了基础。在单个计算节点(32个CPU核心)上,PLQM-VM2计算蛋白质-配体复合物化学势的基准时间,对于单点能量方法约为30 - 45分钟,对于进行几何优化的剪裁方法约为5小时,对于进行几何优化的完整蛋白质FMO方法约为35小时。