Kimura Kimitoshi, Subramanian Ayshwarya, Yin Zhuoran, Khalilnezhad Ahad, Wu Yufan, He Danyang, Dixon Karen O, Chitta Udbhav Kasyap, Ding Xiaokai, Adhikari Niraj, Guzchenko Isabell, Zhang Xiaoming, Tang Ruihan, Pertel Thomas, Myers Samuel A, Aastha Aastha, Nomura Masashi, Eskandari-Sedighi Ghazaleh, Singh Vasundhara, Liu Lei, Lambden Conner, Kleemann Kilian L, Gupta Neha, Barry Jen-Li, Durao Ana, Cheng Yiran, Silveira Sebastian, Zhang Huiyuan, Suhail Aamir, Delorey Toni, Rozenblatt-Rosen Orit, Freeman Gordon J, Selkoe Dennis J, Weiner Howard L, Blurton-Jones Mathew, Cruchaga Carlos, Regev Aviv, Suvà Mario L, Butovsky Oleg, Kuchroo Vijay K
The Gene Lay Institute of Immunology and Inflammation, Brigham and Women's Hospital, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA.
Ann Romney Center for Neurologic Diseases, Brigham and Women's Hospital and Harvard Medical School, Boston, MA, USA.
Nature. 2025 May;641(8063):718-731. doi: 10.1038/s41586-025-08852-z. Epub 2025 Apr 9.
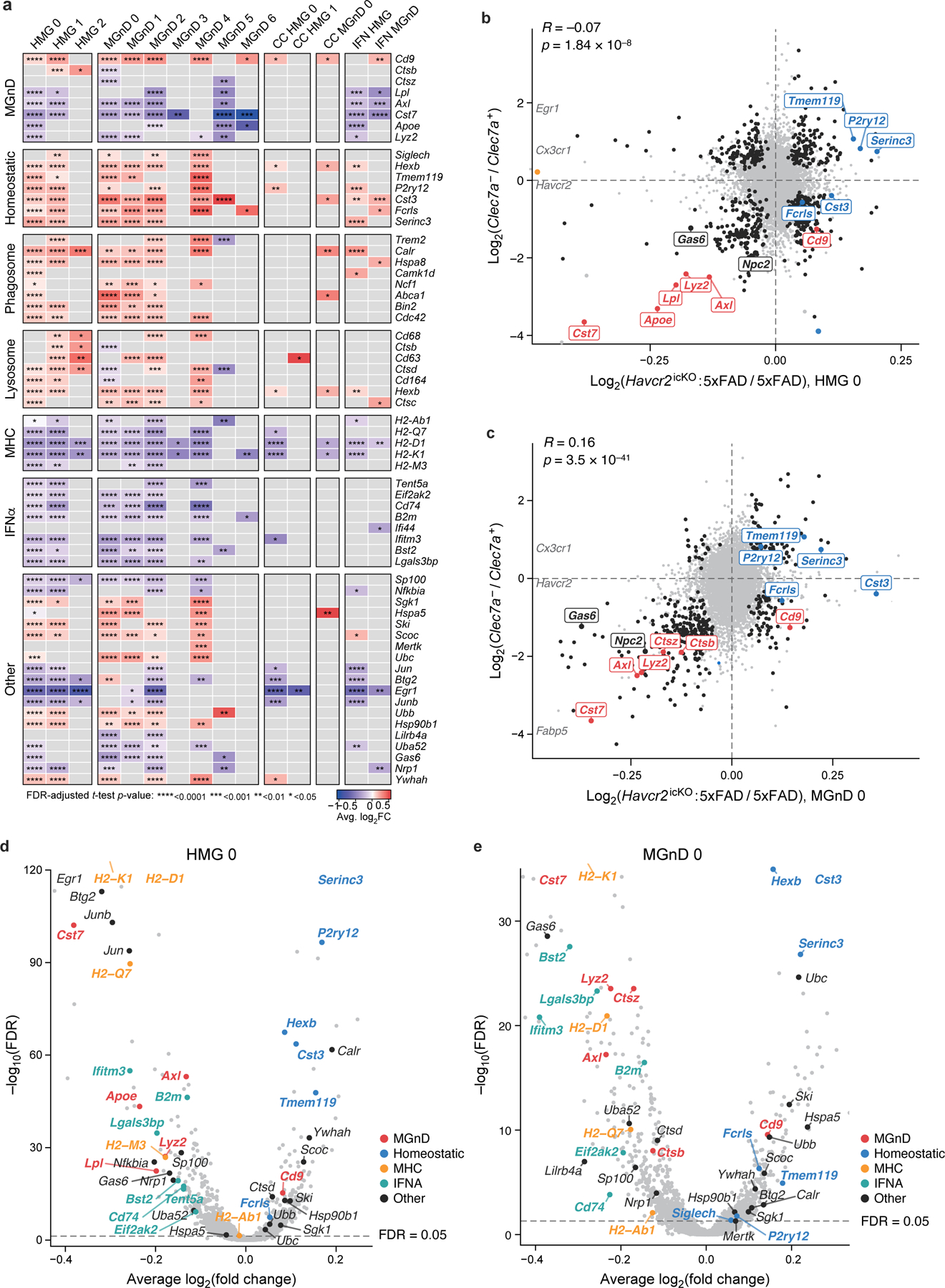
Microglia are the resident immune cells in the brain and have pivotal roles in neurodevelopment and neuroinflammation. This study investigates the function of the immune-checkpoint molecule TIM-3 (encoded by HAVCR2) in microglia. TIM-3 was recently identified as a genetic risk factor for late-onset Alzheimer's disease, and it can induce T cell exhaustion. However, its specific function in brain microglia remains unclear. We demonstrate in mouse models that TGFβ signalling induces TIM-3 expression in microglia. In turn, TIM-3 interacts with SMAD2 and TGFBR2 through its carboxy-terminal tail, which enhances TGFβ signalling by promoting TGFBR-mediated SMAD2 phosphorylation, and this process maintains microglial homeostasis. Genetic deletion of Havcr2 in microglia leads to increased phagocytic activity and a gene-expression profile consistent with the neurodegenerative microglial phenotype (MGnD), also referred to as disease-associated microglia (DAM). Furthermore, microglia-targeted deletion of Havcr2 ameliorates cognitive impairment and reduces amyloid-β pathology in 5×FAD mice (a transgenic model of Alzheimer's disease). Single-nucleus RNA sequencing revealed a subpopulation of MGnD microglia in Havcr2-deficient 5×FAD mice characterized by increased pro-phagocytic and anti-inflammatory gene expression alongside reduced pro-inflammatory gene expression. These transcriptomic changes were corroborated by single-cell RNA sequencing data across most microglial clusters in Havcr2-deficient 5×FAD mice. Our findings reveal that TIM-3 mediates microglia homeostasis through TGFβ signalling and highlight the therapeutic potential of targeting microglial TIM-3 in Alzheimer's disease.
小胶质细胞是大脑中的常驻免疫细胞,在神经发育和神经炎症中起关键作用。本研究调查了免疫检查点分子TIM-3(由HAVCR2编码)在小胶质细胞中的功能。TIM-3最近被确定为晚发性阿尔茨海默病的遗传风险因素,并且它可以诱导T细胞耗竭。然而,其在脑小胶质细胞中的具体功能仍不清楚。我们在小鼠模型中证明,TGFβ信号传导诱导小胶质细胞中TIM-3的表达。反过来,TIM-3通过其羧基末端尾巴与SMAD2和TGFBR2相互作用,通过促进TGFBR介导的SMAD2磷酸化来增强TGFβ信号传导,并且这个过程维持小胶质细胞的稳态。小胶质细胞中Havcr2的基因缺失导致吞噬活性增加以及与神经退行性小胶质细胞表型(MGnD,也称为疾病相关小胶质细胞(DAM))一致的基因表达谱。此外,小胶质细胞靶向缺失Havcr2可改善5×FAD小鼠(阿尔茨海默病的转基因模型)的认知障碍并减少淀粉样β病理。单核RNA测序揭示了Havcr2缺陷型5×FAD小鼠中MGnD小胶质细胞的一个亚群,其特征是促吞噬和抗炎基因表达增加,同时促炎基因表达减少。这些转录组变化在Havcr2缺陷型5×FAD小鼠的大多数小胶质细胞簇的单细胞RNA测序数据中得到了证实。我们的研究结果表明,TIM-3通过TGFβ信号传导介导小胶质细胞稳态,并突出了靶向小胶质细胞TIM-3在阿尔茨海默病中的治疗潜力。