Luppino Federica, Lenz Swantje, Chow Chi Fung Willis, Toth-Petroczy Agnes
Max Planck Institute of Molecular Cell Biology and Genetics, Pfotenhauerstrasse 108, 01307, Dresden, Germany.
Center for Systems Biology Dresden, Pfotenhauerstrasse 108, 01307, Dresden, Germany.
BMC Genomics. 2025 Apr 12;26(1):367. doi: 10.1186/s12864-025-11534-9.
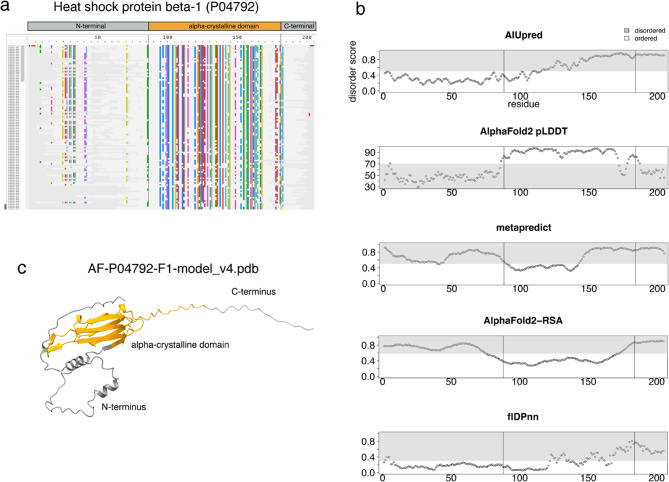
The recent AI breakthrough of AlphaFold2 has revolutionized 3D protein structural modeling, proving crucial for protein design and variant effects prediction. However, intrinsically disordered regions-known for their lack of well-defined structure and lower sequence conservation-often yield low-confidence models. The latest Variant Effect Predictor (VEP), AlphaMissense, leverages AlphaFold2 models, achieving over 90% sensitivity and specificity in predicting variant effects. However, the effectiveness of tools for variants in disordered regions, which account for 30% of the human proteome, remains unclear.
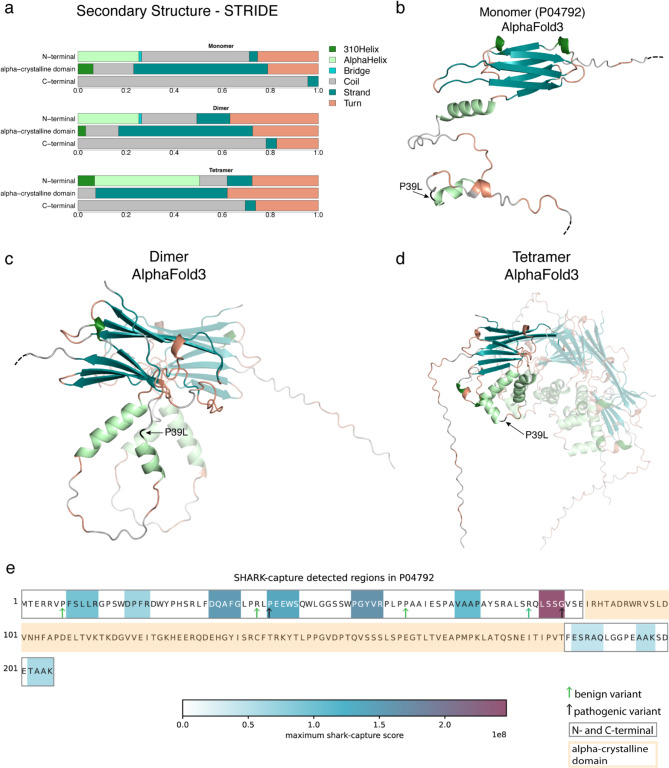
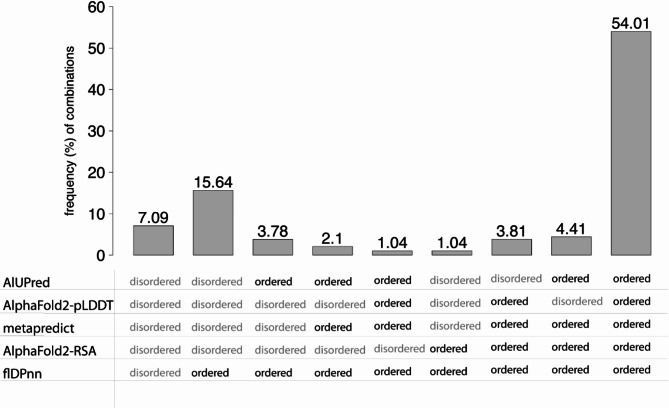
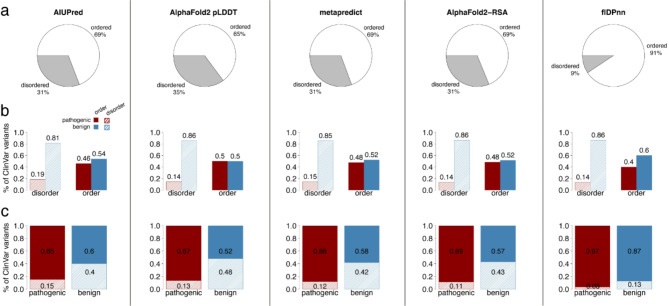
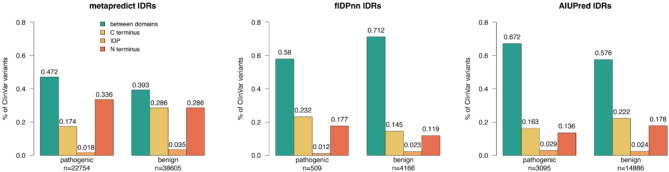
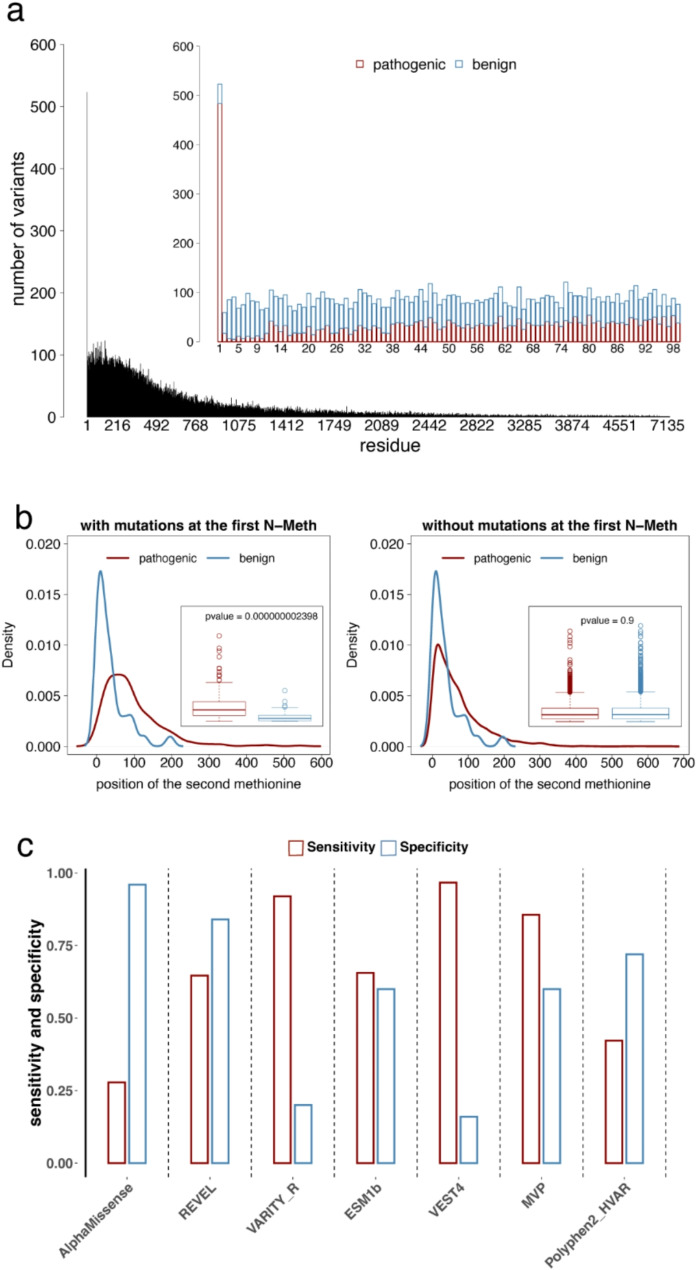
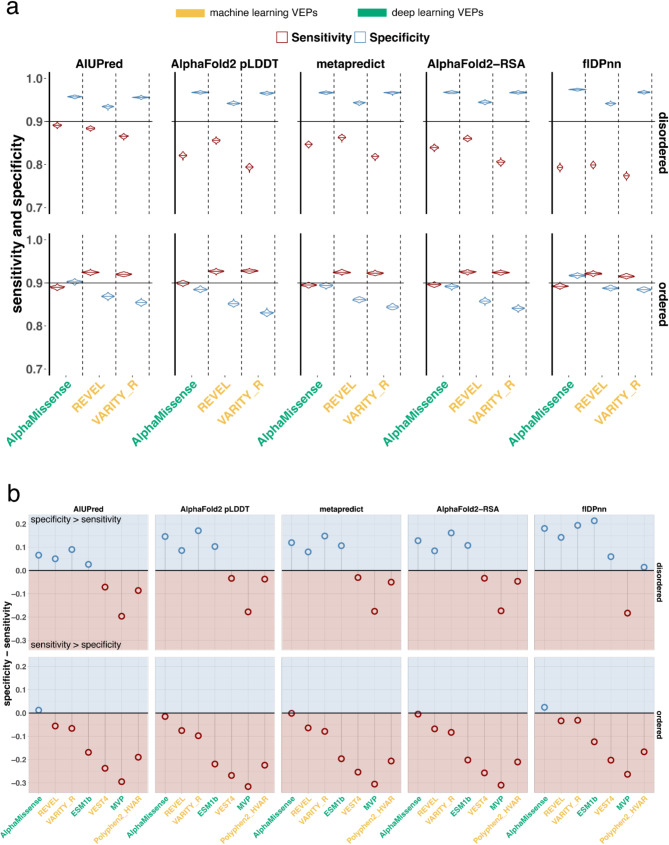
In this study, we found that predicting pathogenicity for variants in disordered regions is less accurate than in ordered regions, particularly for mutations at the first N-Methionine site. Investigations into the efficacy of variant effect predictors on intrinsically disordered regions (IDRs) indicated that mutations in IDRs are predicted with lower sensitivity and the gap between sensitivity and specificity is largest in disordered regions, especially for AlphaMissense and VARITY.
The prevalence of IDRs within the human proteome, coupled with the increasing repertoire of biological functions they are known to perform, necessitated an investigation into the efficacy of state-of-the-art VEPs on such regions. This analysis revealed their consistently reduced sensitivity and differing prediction performance profile to ordered regions, indicating that new IDR-specific features and paradigms are needed to accurately classify disease mutations within those regions.
最近AlphaFold2在人工智能方面的突破彻底改变了3D蛋白质结构建模,这对蛋白质设计和变异效应预测至关重要。然而,内在无序区域因其缺乏明确的结构和较低的序列保守性,往往会产生低置信度的模型。最新的变异效应预测器(VEP)AlphaMissense利用AlphaFold2模型,在预测变异效应方面实现了超过90%的灵敏度和特异性。然而,对于占人类蛋白质组30%的无序区域中的变异,工具的有效性仍不明确。
在本研究中,我们发现预测无序区域中变异的致病性比有序区域更不准确,特别是对于第一个N-甲硫氨酸位点的突变。对变异效应预测器在内在无序区域(IDR)上的功效进行的研究表明,IDR中的突变预测灵敏度较低,且在无序区域中灵敏度与特异性之间的差距最大,尤其是对于AlphaMissense和VARITY。
人类蛋白质组中IDR的普遍性,以及已知它们执行的生物功能种类不断增加,使得有必要研究最先进的VEP在此类区域上的功效。该分析揭示了它们在无序区域中灵敏度持续降低,且预测性能与有序区域不同,这表明需要新的IDR特异性特征和范式来准确分类这些区域内的疾病突变。