Bouvet Delphine, Blondel Amélie, de Sainte Agathe Jean-Madeleine, Leroy Gwendoline, Saint-Martin Cécile, Bellanné-Chantelot Christine
Department of Medical Genetics, AP-HP Pitié-Salpêtrière Hospital, Sorbonne University, Paris, France.
Hum Mutat. 2023 Aug 31;2023:6661013. doi: 10.1155/2023/6661013. eCollection 2023.
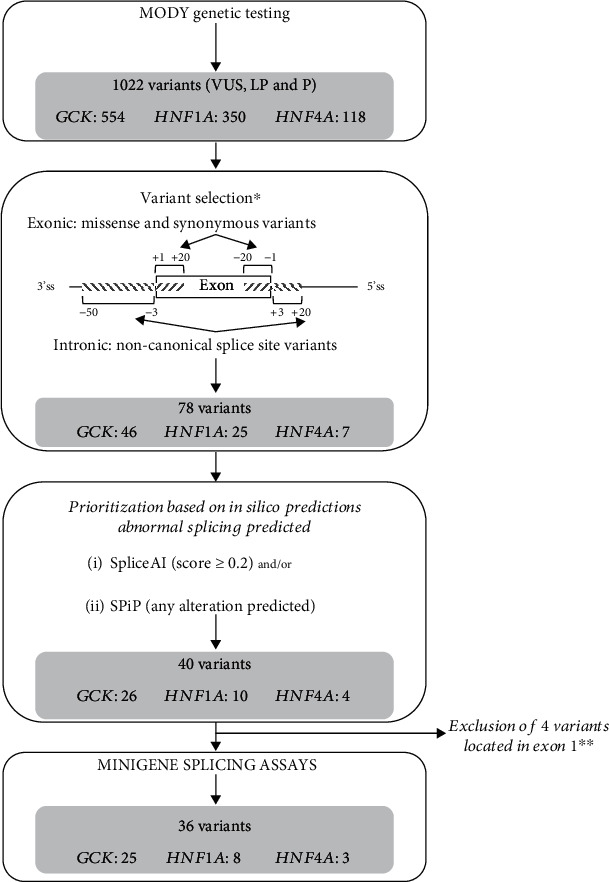
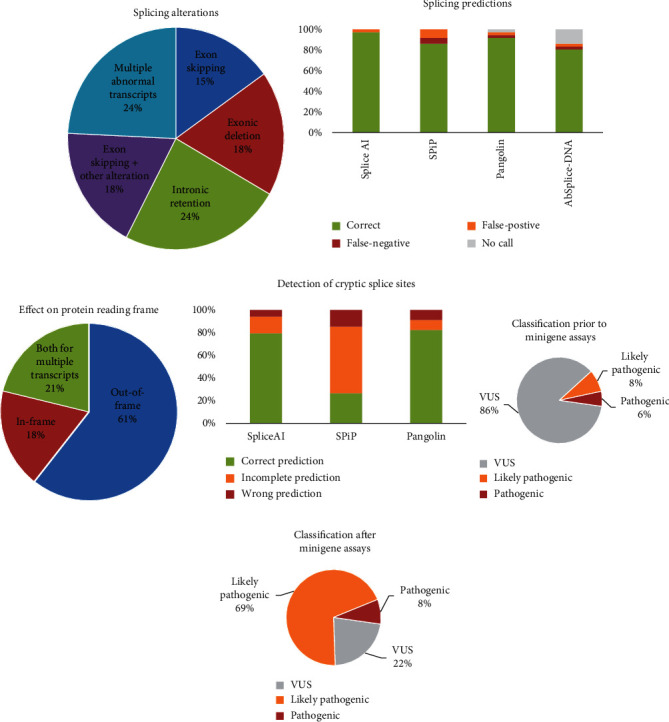
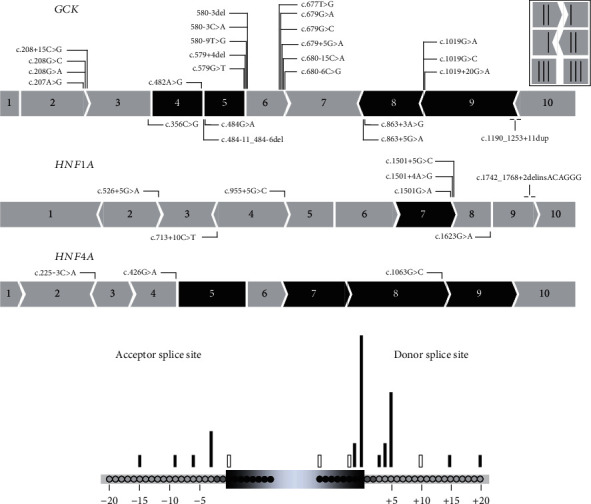
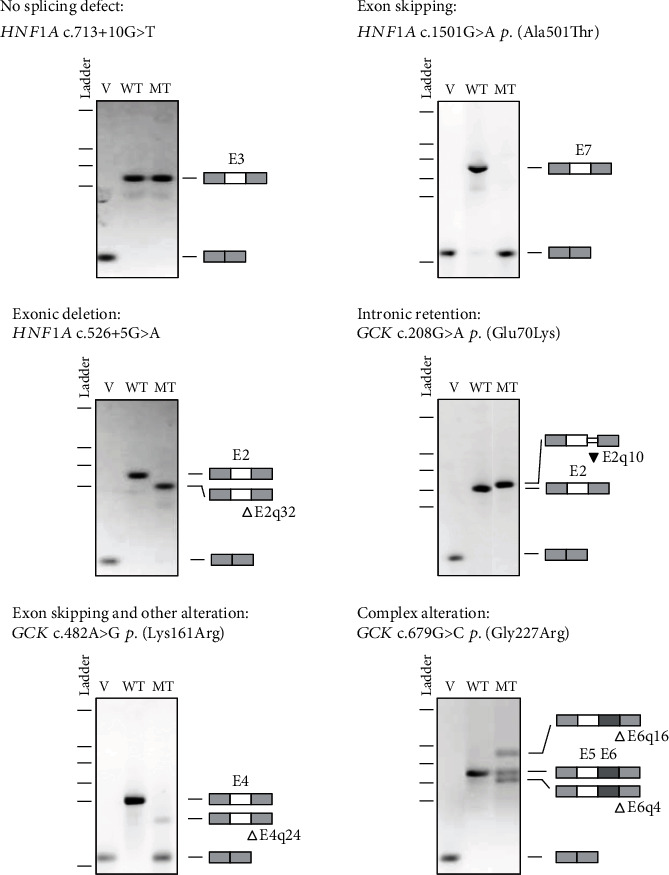
Variants in , , and genes are the three main causes of monogenic diabetes. Determining the molecular etiology is essential for patients with monogenic diabetes to benefit from the most appropriate treatment. The increasing number of variants of unknown significance (VUS) is a major issue in genetic diagnosis, and assessing the impact of variants on RNA splicing is challenging, particularly for genes expressed in tissues not easily accessible as in monogenic diabetes. The in vitro functional splicing assay based on a minigene construct is an appropriate approach. Here, we performed in silico analysis using SpliceAI and SPiP and prioritized 36 spliceogenic variants in , , and . Predictions were secondarily compared with Pangolin and AbSplice-DNA bioinformatics tools which include tissue-specific annotations. We assessed the effect of selected variants on RNA splicing using minigene assays. These assays validated splicing defects for 33 out of 36 spliceogenic variants consisting of exon skipping (15%), exonic deletions (18%), intronic retentions (24%), and complex splicing patterns (42%). This provided additional evidence to reclassify 23 out of 31 (74%) VUS including missense, synonymous, and intronic noncanonical splice site variants as likely pathogenic variants. Comparison of in silico analysis with minigene results showed the robustness of bioinformatics tools to prioritize spliceogenic variants, but revealed inconsistencies in the location of cryptic splice sites underlying the importance of confirming predicted splicing alterations with functional splicing assays. Our study underlines the feasibility and the benefits of implementing minigene-splicing assays in the genetic testing of monogenic diabetes after a prior in-depth in silico analysis.
、和基因的变异是单基因糖尿病的三个主要病因。确定分子病因对于单基因糖尿病患者从最合适的治疗中获益至关重要。意义未明变异(VUS)数量的不断增加是基因诊断中的一个主要问题,评估变异对RNA剪接的影响具有挑战性,特别是对于像单基因糖尿病中那样在不易获取的组织中表达的基因。基于小基因构建体的体外功能剪接分析是一种合适的方法。在这里,我们使用SpliceAI和SPiP进行了计算机分析,并对、和中的36个剪接变异进行了优先级排序。其次,将预测结果与包括组织特异性注释的Pangolin和AbSplice-DNA生物信息学工具进行了比较。我们使用小基因分析评估了所选变异对RNA剪接的影响。这些分析验证了36个剪接变异中的33个存在剪接缺陷,包括外显子跳跃(15%)、外显子缺失(18%)、内含子保留(24%)和复杂剪接模式(42%)。这为将31个(74%)VUS中的23个重新分类提供了额外证据,这些VUS包括错义、同义以及内含子非规范剪接位点变异,将其重新分类为可能的致病变异。计算机分析与小基因结果的比较显示了生物信息学工具在对剪接变异进行优先级排序方面的稳健性,但也揭示了潜在隐匿剪接位点位置的不一致性,这凸显了通过功能剪接分析来确认预测的剪接改变的重要性。我们的研究强调了在单基因糖尿病基因检测中,在进行深入的计算机分析之后实施小基因剪接分析的可行性和益处。