Liu Zhiwei, Liu Pu, Sun Yingying, Nie Zongxiang, Zhang Xiaofan, Zhang Yuqi, Chen Yi, Guo Tiannan
Affiliated Hangzhou First People's Hospital, State Key Laboratory of Medical Proteomics, School of Medicine, Westlake University, Hangzhou, Zhejiang Province, China.
Westlake Center for Intelligent Proteomics, Westlake Laboratory of Life Sciences and Biomedicine, Hangzhou, Zhejiang Province, China.
Nat Commun. 2025 Apr 14;16(1):3530. doi: 10.1038/s41467-025-58866-4.
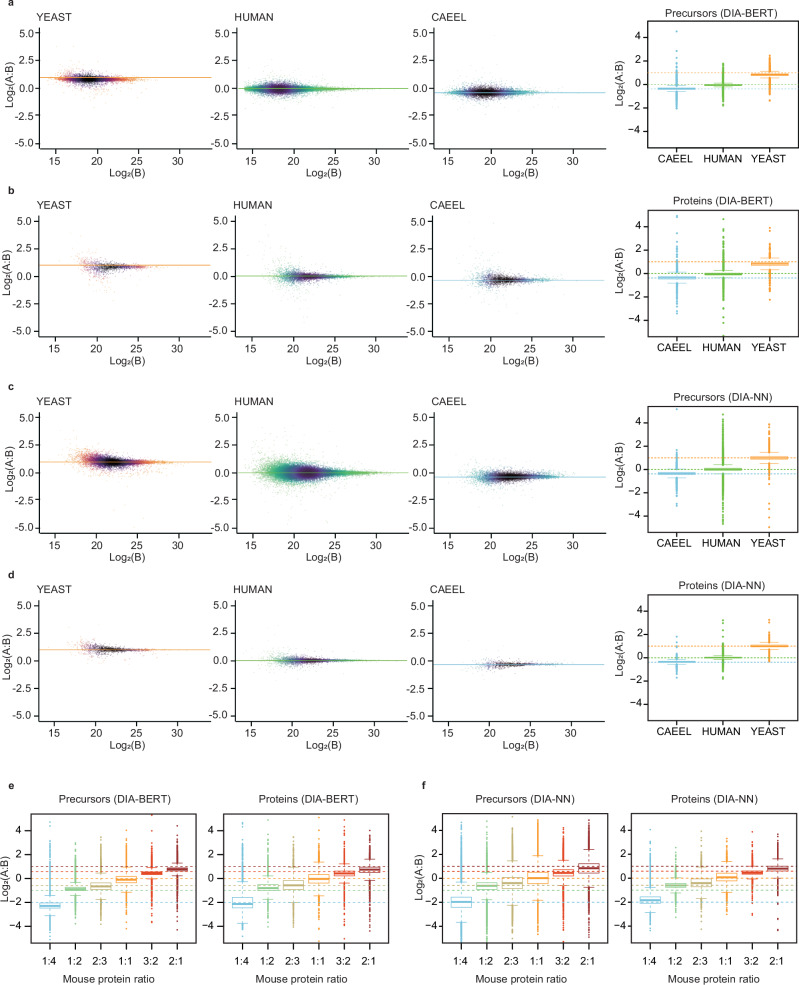
Data-independent acquisition mass spectrometry (DIA-MS) has become increasingly pivotal in quantitative proteomics. In this study, we present DIA-BERT, a software tool that harnesses a transformer-based pre-trained artificial intelligence (AI) model for analyzing DIA proteomics data. The identification model was trained using over 276 million high-quality peptide precursors extracted from existing DIA-MS files, while the quantification model was trained on 34 million peptide precursors from synthetic DIA-MS files. When compared to DIA-NN, DIA-BERT demonstrated a 51% increase in protein identifications and 22% more peptide precursors on average across five human cancer sample sets (cervical cancer, pancreatic adenocarcinoma, myosarcoma, gallbladder cancer, and gastric carcinoma), achieving high quantitative accuracy. This study underscores the potential of leveraging pre-trained models and synthetic datasets to enhance the analysis of DIA proteomics.
数据非依赖型采集质谱法(DIA-MS)在定量蛋白质组学中变得越来越关键。在本研究中,我们展示了DIA-BERT,这是一种软件工具,它利用基于Transformer的预训练人工智能(AI)模型来分析DIA蛋白质组学数据。识别模型使用从现有DIA-MS文件中提取的超过2.76亿个高质量肽前体进行训练,而定量模型则使用来自合成DIA-MS文件的3400万个肽前体进行训练。与DIA-NN相比,在五个人类癌症样本集(宫颈癌、胰腺腺癌、肉瘤、胆囊癌和胃癌)中,DIA-BERT的蛋白质识别率平均提高了51%,肽前体数量平均增加了22%,实现了高定量准确性。这项研究强调了利用预训练模型和合成数据集来加强DIA蛋白质组学分析的潜力。