Joo Jihoon E, Viana-Errasti Julen, Buchanan Daniel D, Valle Laura
Colorectal Oncogenomics Group, Department of Clinical Pathology, The University of Melbourne, Parkville, VIC, Australia.
Collaborative Centre for Genomic Cancer Medicine, Victorian Comprehensive Cancer Centre, Parkville, VIC, Australia.
Fam Cancer. 2025 Apr 16;24(2):38. doi: 10.1007/s10689-025-00460-0.
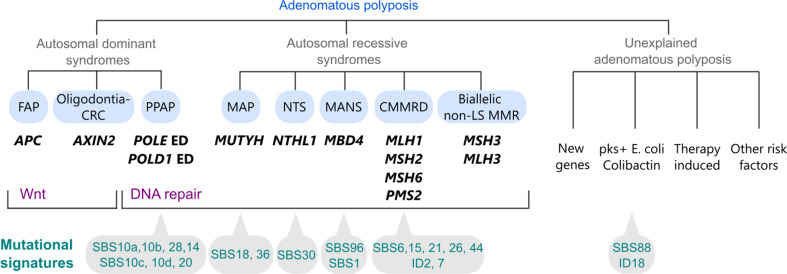
Adenomatous polyposis syndromes are hereditary conditions characterised by the development of multiple adenomas in the gastrointestinal tract, particularly in the colon and rectum, significantly increasing the risk of colorectal cancer and, in some cases, extra-colonic malignancies. These syndromes are caused by germline pathogenic variants (PVs) in genes involved in Wnt signalling and DNA repair. The main autosomal dominant adenomatous polyposis syndromes include familial adenomatous polyposis (FAP) and polymerase proofreading-associated polyposis (PPAP), caused by germline PVs in APC and the POLE and POLD1 genes, respectively. Autosomal recessive syndromes include those caused by biallelic PVs in the DNA mismatch repair genes MLH1, MSH2, MSH6, PMS2, MSH3 and probably MLH3, and in the base excision repair genes MUTYH, NTHL1 and MBD4. This review provides an in-depth discussion of the genetic and molecular mechanisms underlying hereditary adenomatous polyposis syndromes, their clinical presentations, tumour mutational signatures, and emerging approaches for the treatment of the associated cancers. Considerations for genetic testing are described, including post-zygotic mosaicism, non-coding PVs, the interpretation of variants of unknown significance and cancer risks associated with monoallelic variants in the recessive genes. Despite advances in genetic testing and the recent identification of new adenomatous polyposis genes, many cases of multiple adenomas remain genetically unexplained. Non-genetic factors, including environmental risk factors, prior oncologic treatments, and bacterial genotoxins colonising the intestine - particularly colibactin-producing Escherichia coli - have emerged as alternative pathogenic mechanisms.
腺瘤性息肉病综合征是遗传性疾病,其特征是胃肠道,尤其是结肠和直肠中出现多个腺瘤,显著增加了患结直肠癌的风险,在某些情况下还会增加结肠外恶性肿瘤的风险。这些综合征是由参与Wnt信号传导和DNA修复的基因中的种系致病性变异(PVs)引起的。主要的常染色体显性腺瘤性息肉病综合征包括家族性腺瘤性息肉病(FAP)和聚合酶校对相关息肉病(PPAP),分别由APC基因以及POLE和POLD1基因中的种系PVs引起。常染色体隐性综合征包括由DNA错配修复基因MLH1、MSH2、MSH6、PMS2、MSH3以及可能的MLH3,以及碱基切除修复基因MUTYH、NTHL1和MBD4中的双等位基因PVs引起的综合征。本综述深入讨论了遗传性腺瘤性息肉病综合征的遗传和分子机制、其临床表现、肿瘤突变特征以及相关癌症的新兴治疗方法。描述了基因检测的注意事项,包括合子后镶嵌现象、非编码PVs、意义未明变异的解释以及与隐性基因中单等位基因变异相关的癌症风险。尽管基因检测取得了进展,并且最近发现了新的腺瘤性息肉病基因,但许多多发性腺瘤病例在遗传上仍无法解释。非遗传因素,包括环境风险因素、先前的肿瘤治疗以及定植于肠道的细菌基因毒素——尤其是产大肠杆菌素的大肠杆菌——已成为替代致病机制。